Hi,
We’re using ITS2 to examine the fungal community in pig fecal samples, based on this workflow Fungal ITS analysis tutorial, ITS3/4 primers, and the UNITE Fungi plus other Eukaryotes database. We removed unassigned sequences but would like to include the assigned non-fungal ASVs (host, plants, other microbial eukaryotes), in our downstream analyses as we are interested in their biology. However, it’s recommended to remove these non-fungal ASVs. We did find mention of including them in this post but have further questions. Should nonfungi microorganisms be removed from Mycobiome analysis? - #3 by SoilRotifer
If ASVs assigned to non-fungi are true ITS2 sequences and not just off target amplifications, is it incorrect to include them in downstream analyses? Since ITS2 has also been used in plants, animals, and protists as a barcode (albeit with different primers), it seems that these sequences could be viable if validated with a database which is more populated than UNITE for non-fungal taxa (for example using blast with NCBI-nr) and interpreted with care. Since these sequences were amplified with primers developed for fungi, they would not represent a comprehensive set of non-fungal taxa, but could they still be recognized as a subset of eukaryotes other than fungi present in the samples?
If these non-fungal taxa are removed, as they often are, could this bias the relative abundances of fungal taxa?
Thank you!
Indeed, in a typical ITS analysis of fungal communities (as in the tutorial that you mentioned), non-fungal features should be filtered out. Because if you are looking at the mycobiome, plants, animals etc are non-target and not part of the microbial community.
But in your case you are not only looking at the mycobiome, you are doing a diet metabarcoding analysis as well.
So for your case as you want two analyses for the price of one I recommend: (1) filtering out non-fungal non-microbial features for mycobiome analysis; (2) in a separate analysis filtering out all microbial fungal features for diet analysis (keeping mushrooms, which would be dietary).
Yes, because these non-fungal ASVs are separate from the microbial community that you are trying to describe; they are not living, interacting members of the gut mycobiome but instead are dietary constituents. And similarly, intestinal fungi should be removed from the dataset for diet metabarcoding analysis because these are (presumably) not intentional dietary constituents so if you are trying to describe what an animal is eating these ASVs should be removed. It is about cleanly separating these constituents to describe different compartments (dietary intake vs. gut mycobiome community).
If they are not removed it will bias the relative abundances of fungal taxa, as relative abundance could be highly skewed by differences in diet. If they are removed, it should not create a bias as long as sufficient fungal reads remain and you normalize appropriately afterwards.
I have not used it yet so cannot share my experience with it, but Eukaryome seems to fit the bill. They even compile QIIME 2-compatible files: https://eukaryome.org/qiime2/
and other forums users have kindly provided pre-trained naive Bayes classifiers of this database:
I recommend checking this. If you can acquire a reliable reference set of non-fungal ITS sequences (e.g., eukaryome, and filter out the fungi) you could use a primer evaluation tool to see how much coverage etc your primers have of different groups.
Sorry about my delayed reply, and thanks very much for your helpful discussion! I will definitely check out Eukaryome and thank you for the suggestion of splitting the dataset into dietary and fungal components. A couple follow up questions if you don’t mind:
What is the best way to interpret non-fungal taxon abundances? Since binding efficiency of ITS3/4 is optimized for fungi, efficiency is likely variable and/or lower for non-fungal taxa (although I need to check this in the literature). Due to variable primer efficiencies, could it be incorrect to compare relative abundances of non-fungal taxa within a sample? However, assuming the factors which cause the variability are similar (or at least random) across treatment groups, I think it would be correct to compare relative abundances of non-fungal taxa across treatment groups?
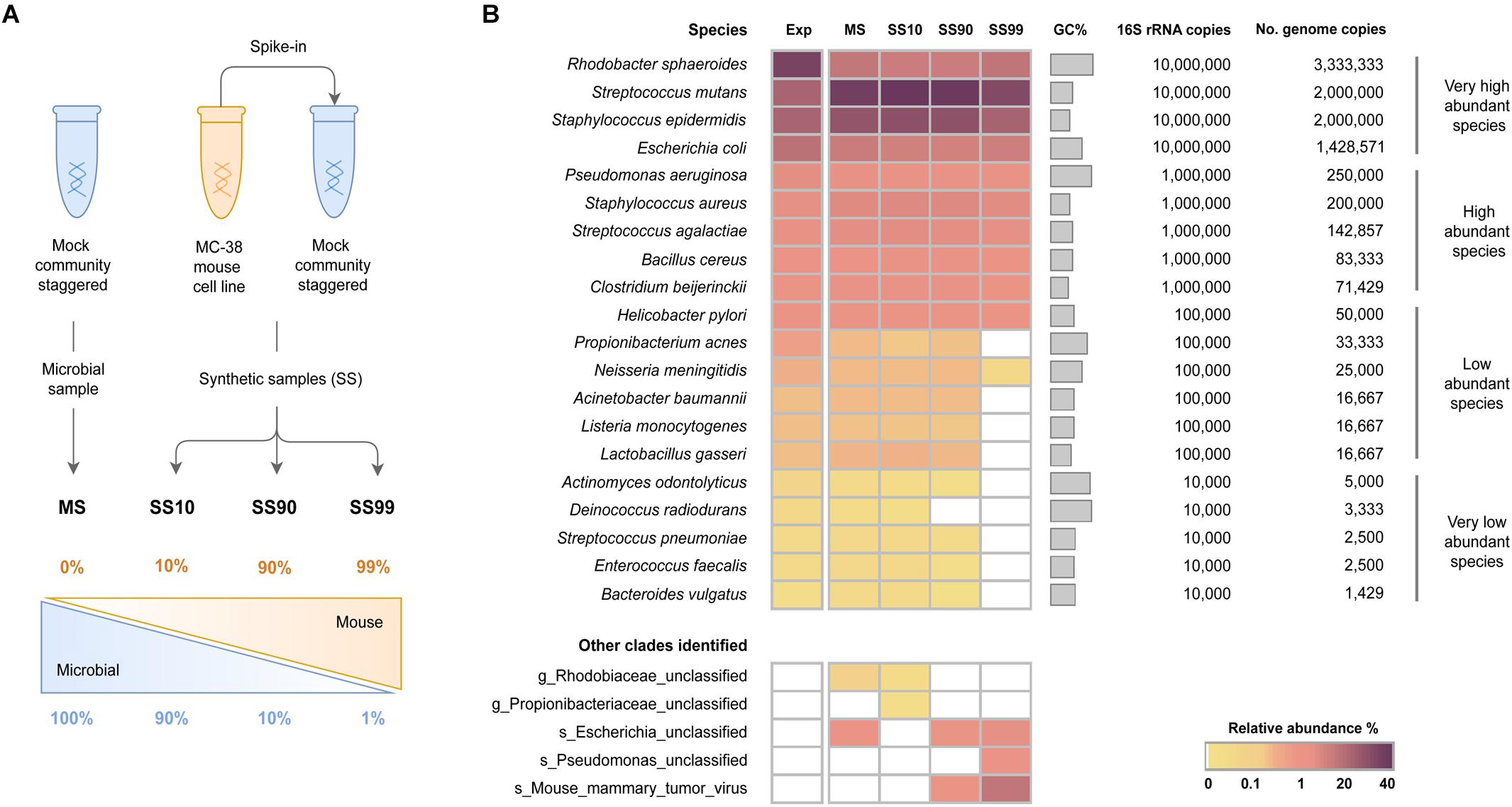
It makes sense that including host DNA (and other non-fungal reads) in downstream analyses could skew relative abundance values, but is this always the case? In studies which use relative abundance data as a proxy for absolute abundance data, are there some cases where excluding host and/or non-fungal reads could bias fungal relative abundances to make them less closely approximate absolute abundances? I think Periera-Marques et al. Frontiers | Impact of Host DNA and Sequencing Depth on the Taxonomic Resolution of Whole Metagenome Sequencing for Microbiome Analysis Figure 1 https://www.frontiersin.org/files/Articles/454372/fmicb-10-01277-HTML/image_m/fmicb-10-01277-g001.jpg illustrates this possibility. Here, the authors profile synthetic shotgun metagenomic samples with varying levels of host DNA spiked into a bacterial mock community - SS10 (10% host DNA) to SS99 (99% host DNA). (I have ITS2 data, but I think the concepts are similar). In this figure, the relative abundances of each taxon across samples (SS10, SS90, SS99) are understandably similar, but a plot of absolute abundances would look very different. Wouldn’t a relative abundance plot of the data which includes the host DNA better approximate absolute abundances here (not useful for this paper – which has a different goal – but just as an example)? This is an extreme case, as the host and microbial loads vary greatly. But, in real samples where the microbial and/or host loads do vary greatly across sample, could it be advantageous to leave in host DNA in downstream relative abundance analyses to best approximate absolute abundances? (I realize lab/bioinformatic methods would be best to use to correct for compositionality/best approximate absolute abundances, but I’m still curious about this question in the context of removing host from downstream analyses).
These are great questions! I'm glad you are thinking deeply about how the primers work and their limitations.
You're right to be cautious. Directly comparing non-fungal relative abundances from ITS data is problematic due to the primer bias.
Correct! If the bias is consistent, then comparison may be possible.
Zooming out, this is a problem with amplicons generally. It may be especially bad for amplicons detected as an off-target effect from another region.
On the topic of relative vs absolute abundance:
Wouldn’t a relative abundance plot of the data which includes the host DNA better approximate absolute abundances here?
to best approximate absolute abundances
Yes, I suppose you can choose what you include in the denominator of the fraction!
x/total reads
x/non-host reads
x/fungal reads
In shotgun sequencing, where everything is captured, I can see the value of using 'total reads' as the denominator. But amplicons are not unbiased! We are choosing primers to amplify regions that capture diversity we care about!
So... I've attempted to use qPCR to calculate absolute abundance from amplicons. If you have an external data source you can use for normalization, you could try something similar.
{kind=link}