After importing my multiplexed files from an ITS MiSeq 2 x 300 sequencing, I am encountering the same message reported in this topic at position 37 for both R1 and R2.
I understand that, in their case, it was a bug from a previous version but I was wondering what would it mean in this case. Consequently, I'm losing many reads when filtered with dada2, which didn't happen in a previous sequencing run with the exact same library prep.
Thank you so much for searching the forum before posting! That is such a huge help for us . Unfortunately, that error report you linked to is not applicable here (please allow me to explain!). In that issue, the bug (or error) was in the values of the message itself - "This position (249) is greater than the minimum sequence length observed during subsampling (251 bases)". According to my calculations, 249 is not less than 251 (not greater than), which is why that error was probably reported in the first place.
Okay, so on to your data. This isn't a bug , or even an error, it is just a cautionary warning to you:
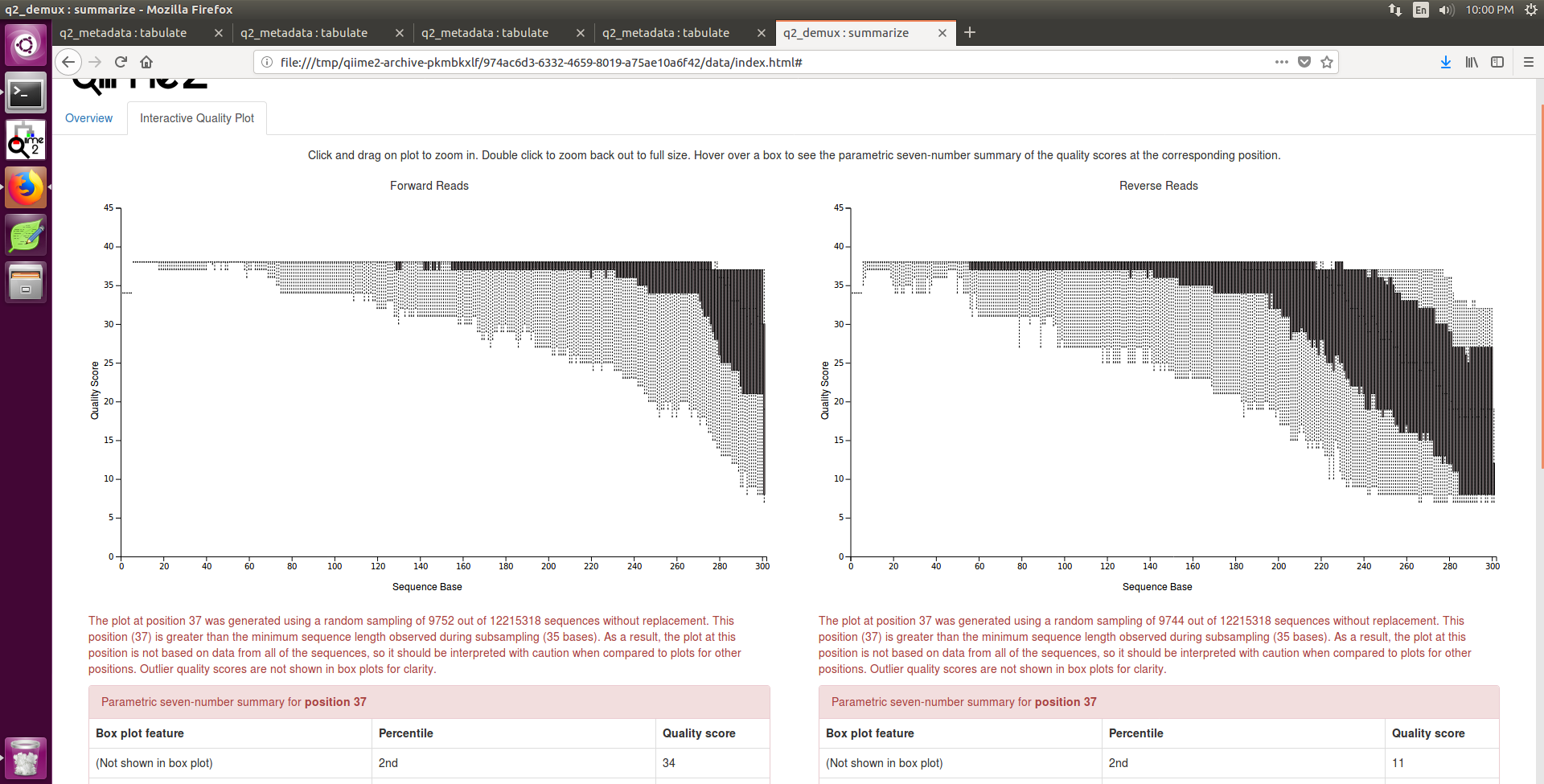
This position (37) is greater than the minimum sequence length observed during subsampling (35 bases)
What that means is that the shortest read observed in these sequences was 35nts long. The reason there is a warning here is because the number of observations represented in the boxplot at this position is less than the number of reads present at earlier bp positions. That is the kind of thing to keep in mind when comparing the boxplots --- it isn't exactly a fair comparison if the "sample size" for one box plot is different from another.
Another way to look at this is to check out the "Demultiplexed sequence length summary" --- the low end of your read distribution is only 35 nts long , while it quickly jumps up to ~210ish nts. If all of your reads were the same length you would see the same number for each percentile in this 7-number summary.
Well, it looks like you have a wide spread of read lengths --- it is possible that the trim/trunc params your are running DADA2 with might not provide sufficient overlap when merging the reads (DADA2 needs ~20nt overlap). Have you tried running just your forward reads through q2-dada2?
I'm actually running qiime dada2 denoise-single \ and with --trunc 285 and --trim 0 since in previous analyses I was losing many seqs at merging. Any recommendations on how to increase the number of retained seqs during dada2?
Have you had a chance to read through the dada2 docs yet? If not, I highly recommend it. Otherwise, just eyeballing that screenshot above, I wonder what a trunc of ~270-275 would do for you?
trunc of 270 helped on keeping a larger portion of reads after dada2
I still keep a slightly lower percentage in this run compared to past runs with exact same prep, but might just be the specific run. Is there an adequate percentage of reads that should be kept? Seems like many ~35-45% are dropped in the filtering step of dada2 for this run.
35-45% is not bad at all. There is no "normal", of course, since it is going to vary based on run quality. But I think there have been some posts on this forum where the consensus has been that a 75% read loss on that user's data is not outrageous.
It is more important to check where the sequences are being lost. If at the filtering/denoising stage, there is not much you can do (you can adjust filtering and trimming parameters to see how that helps; trimming more low-quality bases in particular can help a lot). If at the merging stage, you should carefully review read lengths, amplicon length distributions, and trimming parameters before deciding if this is "bad" and what you should do about it. There are lots of posts on this forum discussing these topics that you can review for more advice.