![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Citations:
If you use the following COI resources or RESCRIPt for COI database preparation, please cite the following:
-
Michael S Robeson II, Devon R O’Rourke, Benjamin D Kaehler, Michal Ziemski, Matthew R Dillon, Jeffrey T Foster, Nicholas A Bokulich. RESCRIPt: Reproducible sequence taxonomy reference database management for the masses. bioRxiv 2020.10.05.326504; doi: https://doi.org/10.1101/2020.10.05.326504
-
O'Rourke, DR, Bokulich, NA, Jusino, MA, MacManes, MD, Foster, JT. A total crapshoot? Evaluating bioinformatic decisions in animal diet metabarcoding analyses. Ecol Evol. 2020; 00: 1– 19. https://doi.org/10.1002/ece3.6594
Preamble
This tutorial is a spinoff of the amazing work done by the developers behind RESCIPRt, and uses many of the same steps illustrated in the tutorial. Shout out to @BenKaehler, @thermokarst @Nicholas_Bokulich, and @SoilRotifer (and anyone else I missed - just let me know!)
Much like the RESCRIPt tutorial, the aim of this document is to provide an example of how to build and evaluate a reference database - but unlike the other RESCRIPt tutorial, we'll focus our attention on Cytochrome Oxidase I (COI) sequence data.
Available resources
Don't want to bother with creating your own database? I hear you... Or, maybe you do want to give it a shot, but can't be bothered to download all the raw data? Yep, that's also a pain. Tired of long tutorials!? Too bad - keep reading! For those of you who want to get your hands on some data right away, I've placed a few different files that you can use to either (1) build your own database from raw BOLD sequences, or (2) use directly in your classification steps in a QIIME 2 environment.
- Sequence data, taxonomy data, and associated metadata were acquired from The Barcode of Life Data System BOLD between July 1, 2020 (happy Canada Day!) and August 8, 2020 (happy International Cat Day!). Information on the process used to gather the data is documented below.
- BOLD sequences were saved as
bold_rawSeqs.qza, available here - BOLD taxonomy data was saved as
bold_rawTaxa.qza, available here - Associated metadata is also available, saved as
bold_rawMetadata_forQiime.tsv.gz, available here
Important caveat #1: these raw data contain all of the publicly available COI sequences I could scrape, so it's highly advised that you filter these before using them to classify you data. I've provided a number of examples of the kind of filtering considerations to apply in this tutorial, but you might find yourself wanting to quickly reduce this massive dataset by filtering for particular taxa, or perhaps using the metadata and filtering for particular country of origins specimen were collected. However you choose to use these raw data, I would strongly encourage you to filter them first prior to classifying anything - it'll improve the quality of classification, and speed it up to boot.
2. Or maybe you couldn't be bothered to filter it all yourself either! I've got you covered - just remember to throw some good karma to your friendly COI database creator, okay? The raw BOLD sequences were initially filtered for ambiguous nucleotide content (5 or more N's), long homopolymer runs (12 or more), very short (< 250 bp) or very long (> 1600 bp) sequences, and dereplicated. This produced a pair of files with the prefix bold_derep1:
- The sequence artifact
bold_derep1_seqs.qzais available here - The taxonomy artifact
bold_derep1_taxa.qzais available here
You might be inclined to use those sequences to classify your COI data, however it's likely that trimming those references to contain only the sequence region spanning your sequence data itself would provide a better classification estimate. In other words, while these reference sequences are often between 500-650 bp, the primers you've used to generate your COI reads are probably not spanning that length. If, for example, you used a primer pair that is a subset of that region, you should follow the section in the tutorial below that shows how to take these bold_derep1 files and createte that primer-specific reference dataset.
I've included one such example of a primer-trimmed COI database. This one is specific to the ANML primers we use in our lab for bird and bat diet analyses. Important caveat #2: don't use these artifact files to classify your COI sequences if you used a different primer set! These primer-trimmed files include:
- The sequence artifact
bold_anml_seqs.qzaavailable here - The sequence artifact
bold_anml_taxa.qzaavailable here
In addition, I created two naive Bayes classifier objects: (1) using all references from the bold_anml data, and (2) a classifier using only Arthropod references from the bold_full data. If you want to classify your sequences with sklearn, you can use the following files, but be aware that what these references do and do not contain with respect to sequence composition and reference taxonomy information ![]() :
:
- The
bold_anml_classifier.qzafile is available here - The
bold_full_ArthOnly_classifier.qzafile is hosted it here.
As with the raw BOLD data, these bold_derep and bold_anml sequence and taxonomy artifacts consist of BOLD records for Animal, Protist, and Fungi taxa. You can certainly modify these by filtering for certain taxonomic groups or other metadata.
Background
Cytochrome Oxidase I is a commonly studied marker gene among animals (and other macro and microeukaryotes including plants, fungi, and protists!). The Barcode of Life Data System BOLD is one such repository of COI sequence information. Unfortunately the last versioned release of COI data came in 2015, however BOLD offers a variety of means with which to gather individual COI records. The following tutorial will show how to:
- Obtain BOLD COI sequences and associated taxonomy information
- Reformat these data and import into QIIME
- Filter these data in QIIME using options available via the RESCRIPt plugin
- Evaluate the database using options via RESCRIPt
- Create a naive Bayes classifier using RESCRIPt which can the be used in downstream classification of your particular COI experiment
Gathering data - overview
Gathering data from BOLD is really simple to do for a handful of species or sequences - just navigate to their taxonomy browser, search for some taxa, click on the "public data" icon once the search is complete, then download the combined sequence and taxonomy records in the red "combined" button (as either an .xml or .tsv format). You can, in principle, use this same method to download the entire BOLD database, but it can take between 2-4 days with a decent internet connection (I've tried), and be warned that if you lose signal, you'll start all over again (I have).
Fortunately BOLD offers another option which is scriptable, and thanks to the wonderful Scott Chamberlain and the ROpenSci community, there's a particular R package, the eponymous bold R library, that can be used to access the Barcode of Life Database from the command line. I created an R script, bold_datapull_byGroup.R that selects all COI records from BOLD by pulling all public records matching for all Animal, Fungi, and Protist taxa. Unfortunately you can't create a single term to search all BOLD COI records, as the Barcode of Life's API times out if you search for too many records at one time. As a result, and after a bunch of trial and error, I ended up breaking up these COI data into a number of smaller groups that I then combine outside of R. The output of this R script, for each group, is a pair of files that contain:
- Sequence information and taxonomic information (used to create a fasta file). The sequence data contains gaps, and while these are dealt with once imported into QIIME, note that if you're trying to combine these raw fasta records with other datasets that don't contain gaps, you might run into errors when importing into QIIME when mixing gapped (ex.
AT--GCG) and ungapped strings (ex.ATGCG). - Metadata including the sequenceID, processid, bin_uri, genbank_accession, country, institution_storing records. While the sequenceID represents a unique sequence identifier within BOLD, the processid applies to the larger specimen itself (as BOLD records may contain images, collection information, and other metadata), while the bin_uri record indicates the BOLD BIN number (which may or may not include multiple records with common or distinct sequence and taxonomic information!). I kept these data in my R script because it seemed like those metadata may be useful when filtering the database (more on one such application below), but I leave it up to the user to get creative in how they use this information for their own purposes.
Gathering data - the easy way
After running the R script, the raw data contained in the individual .csv files were combined and filtered to ensure no duplicate records were retained. The raw fasta and taxonomy text files were then converted into QIIME .qza formats. Links to these raw files are listed in the Available resources section above.
Gathering data - the gory details
Start by executing the R script, bold_datapull_byGroup.R. This creates 10 distinct pairs of .csv files ending with seqNtaxa.csv (for sequence and taxonomic information), and meta.csv (for metadata information). When I first got started building a COI database for analyzing bat diets, I figured I should keep only the arthropod COI data (because all my hosts were consuming were spiders and insects). Nevertheless, I learned that incorporating both expected diet COI data as well as unexpected COI sequences can be useful in data filtering. The chordate records, for instance, included bat COI data that helped me remove host sequences. Likewise, I've noticed that when you sample in an open environment it's possible for other things like fungal DNA to get amplified with your primers you thought were specific to a certain taxa. In short, I haven't found a compelling reason to discard COI data when compiling a database purely based on an unexpected taxonomy, but if someone knows of one I'd love to hear it. Notably, we can and will absolutely remove COI records based on other quality control factors (more on that below).
Once the R script is finished (expect somewhere between 4-24 hours to finish), combine all seqNtaxa.csv files and reformat into a fasta-formatted file (expect between ~ 2 minutes to finish!):
cat *.seqNtaxa.csv | \
grep -v 'sequenceID,taxon,nucleotides' | \
awk -F ',' '{OFS="\t"};{print $1";tax="$2, $3}' | \
sed 's/^/>/g' | \
tr '\t' '\n' > bold_allrawSeqs.fasta
There are a total of 3,518,537 sequences - that's a lot of COI data!
I wanted to make sure there wasn't any duplicate BOLD IDs imported by the process (spoiler, there were...). After a bit of troubleshooting (see below), it looks as if these are very rare, and they occur because there are multiple metadata entries with very similar but not identical records. They contain the same exact sequences, the same exact taxonomic strings, but odd differences in other metadata. To check on these curious cases of duplicate sequenceIDs, I first created a list of all the possible duplicate records:
grep '^>' bold_allrawSeqs.fasta | cut -f 1 -d ';' | sed 's/^>//' | sort | uniq -c | awk '{if ($1>1) print $0}' > duplicated_bold_records.txt
There were 41 instances where the BOLD processID contains at least one duplicate. In all 41 instances, the taxonomic information is identical for each, and there are only single duplicate entries. In other words, all instances where a BOLD ID occurs more than once are instances where they occur exactly two times. These aren't cases where we have different taxonomies assigned to the same BOLD ID - good. But I wanted to confirm whether or not these duplicate entries also just entirely duplicate records. In particular, do they contain the same sequence information?
for comp in $(awk '{print $2}' duplicated_bold_records.txt); do
str1=$(grep -w $comp bold_allrawSeqs.fasta -A 1 | grep -v '^>' | grep -v '^--' | head -1);
str2=$(grep -w $comp bold_allrawSeqs.fasta -A 1 | grep -v '^>' | grep -v '^--' | tail -1);
if [ "$str1" == "$str2" ]; then
echo $comp "Strings are equal"
else
echo $comp "Strings are not equal"
fi
done
note the additional "sort | uniq" line added in making the raw fasta file - it removes those 41 entries...
It turns out every one of the 41 duplicated sequence records contain identical sequence strings (phew!). In this case, we can just delete the list of duplicate records. I opted to just rerun the above command generating the raw fasta file once more, and delete out duplicate entries.
cat *.seqNtaxa.csv | \
grep -v 'sequenceID,taxon,nucleotides' | \
sort | uniq | \
awk -F ',' '{OFS="\t"};{print $1";tax="$2, $3}' | \
sed 's/^/>/g' | \
tr '\t' '\n' > bold_allrawSeqs.fasta
Now there are just 3,518,496 sequences, thus the 41 duplicated records have been removed.
One final bit of a quality control check: it turns out that importing these raw data into QIIME will work, but using these data will fail because of some non-IUPAC characters lingering in the sequences themselves. The following script will search through every sequence and determine whether or not it contains a character outside of the accepted IUPAC character set. We then drop any sequence containing those bad sequences, and generate our final file. Specifically, the valid characters are the set: ['T', 'H', 'A', 'D', 'G', 'R', 'C', '.', 'S', 'M', 'B', 'N', '-', 'W', 'V', 'K', 'Y']
## getting the line numbers of the bad sequences
grep -v '^>' bold_allrawSeqs.fasta | grep [^THADGRC\.SMBNWVKY-] -n > badseqs.txt
## reformatting these line numbers to delete
for val in $(cut -f 1 badseqs.txt -d ':'); do
myvar1=$(($val*2))
myvar2=$(($myvar1-1))
echo $myvar2"d"
echo $myvar1"d"
done | tr '\n' ';' | sed 's/.$//' > droplines.txt
## deleting these line numbers
myval=$(cat droplines.txt)
sed -e $myval bold_allrawSeqs.fasta > cleaned_bold_allrawSeqs.fasta
Now there are 3,518,463 sequences remaining; 33 weird sequences were dropped. Not bad!
Finally, I used that cleaned fasta file to generate the pair of fasta and taxonomy text files in the format that QIIME accepts for import:
## First make the fasta file
sed 's/;tax=k.*//' cleaned_bold_allrawSeqs.fasta > bold_rawSeqs_forQiime.fasta
## Next make the taxonomy file
grep '^>' cleaned_bold_allrawSeqs.fasta | sed 's/^>//' | \
awk '{for(i=1;i<2;i++)sub(";","\t")}1' > bold_rawTaxa_forQiime.tsv
We'll now import two of these files as qiime objects for building our COI database:
bold_rawSeqs_forQiime.fastais used to create a sequence .qza artifact (type == FeatureData[AlignedSequence])bold_rawTaxa_forQiime.tsvis used to create the taxonomy .qza artifact associated with the fasta sequences (type == FeatureData[Taxonomy]). Because the raw BOLD sequence data may contain gaps, we'll need to import it slightly different than a standard fasta (that's why we use a--type 'FeatureData[AlignedSequence]'instead of a more typical--type 'FeatureData[Sequence]'):
## import sequence data
qiime tools import \
--input-path bold_rawSeqs_forQiime.fasta \
--output-path bold_rawSeqs.qza \
--type 'FeatureData[AlignedSequence]'
## import taxonomy data
qiime tools import \
--type 'FeatureData[Taxonomy]' \
--input-format HeaderlessTSVTaxonomyFormat \
--input-path bold_rawTaxa_forQiime.tsv \
--output-path bold_rawTaxa.qza
One last thing before we jump into further processing these database .qza files in QIIME. There is metadata containing the country a sequence was collected, as well as it's (potential) GenBank accession ID, BOLD BIN number, etc. generatd from the same bold_datapull_byGroup.R script used to generate sequence and taxonomy information. These are the *meta.csv files. We combine and reformat these metadata files, but once the reformatting is complete as a standard text file, we're ready to use them directly in QIIME without any further importing as some .qza object.
Why bother with these metadata? You might want to remove any COI sequence that lacks a country of origin, or perhaps you want to restrict your database only to those sequences identified from Costa Rica. Maybe you wanted to discard any BOLD sequence mined from GenBank. You can do all of that by using the metadata information and follow the metadata-based filtering documentation in QIIME.
So, how do we create this metadata file?
cat *.meta.csv | \
grep -v 'sequenceID,processid,bin_uri,genbank_accession,country,institution_storing' | \
awk '{for(i=1;i<6;i++)sub(",","\t")}1' | sort -uk1,1 > bold_rawMetadata_forQiime.tsv
You can use the bold_rawMetadata_forQiime.tsv.gz file as you see fit. I'm curious to hear how users might apply it.
RESCRIPt work
At this point I have hopefully shown you how to complete the first two tasks:
- Obtain BOLD COI sequences and associated taxonomy information
- Reformat these data and import into QIIME
The next step is to apply some sensible filters to these raw sequences. While there isn't a single best practice that would be applicable for every users experiment, I think there are a common set of quality controls that most researchers might want to consider:
- removing sequences with a high number of ambiguous sequences (
N) - removing sequences with an excessive number of homopolymers
- removing extremely short sequences
- removing redundant sequences (dereplicating)
For more on each of these considerations (and more!) see the RESCRIPt tutorial using a SILVA 16S dataset.
The first step in our workflow isn't even listed above - we need to remove the gaps in the BOLD sequences. This isn't necessarily a step other users may need to take should they be importing their own data from other sources, but if you have a - anywhere in the sequence itself, we need to remove it prior to building a classifier. Thankfully, RESCRIPt can tackle this. In the command below, we'll remove the gaps (if present) in a sequence. In addition, the --p-min-length parameter gives us an option to remove extremely short sequences. How short is too short? I wasn't sure when I first started this, and that led me to looking at two things:
- What would the distribution of the sequence lengths be of the degapped sequences look like?
- How would these distributions change if I trimmed the leading and trailing ambiguous characters (
N) from the sequences (but leaving any internal ambiguous sites alone for now)?
I set the minimum length to 1 so that I could first obtain these degapped raw sequences (this took about an hour):
qiime rescript degap-seqs \
--i-aligned-sequences bold_rawSeqs.qza \
--p-min-length 1 \
--o-degapped-sequences bold_degapSeqs_min1.qza
Once I had those sequences, I exported the bold_degapSeqs_min1.qza object back to a standard fasta file, then created a frequency table of the untrimmed sequence lengths:
qiime tools export --input-path bold_degapSeqs_min1.qza --output-path degap_tmpdir
grep -v '^>' ./degap_tmpdir/dna-sequences.fasta | awk '{print length}' | sort | uniq -c | sort -k2,2n > seqlength_untrimmed_hist.txt
Then, I repeated that process but first had to remove the leading/trailing ambiguous nucleotides:
grep -v '^>' ./degap_tmpdir/dna-sequences.fasta | \
sed 's/^N*//' | rev | sed 's/^N*//' | \
awk '{print length}' | sort | uniq -c | sort -k2,2n > seqlength_Ntrimmed_hist.txt
Plotting these frequencies below illustrate that the vast majority of our sequences are in the range of about 500-650 bp, but there are instances of very short (as low as 54 bp) and very long (as long as 7659 bp) COI fragments. It also turns out that the leading/trailing N-trimming didn't make a big difference in the overall distribution of fragment sizes.

The table below highlights that nearly all of our data is retained if we apply a filter under 200 bp, whether or not the N's were trimmed initially. As expected, the N-trimming results in a small decrease in the number of sequences retained relative to untrimmed data for each length threshold, but this effect is very minor generally. For example, if we were to filter at a 500 bp threshold length the difference is 1/10th of a percent. In absolute terms, the difference was 285 sequences fewer sequences among the N-trimmed data (3,510,119 untrimmed vs. 3,509,834 trimmed):
| Min_len(bp) | N-trimmed | untrimmed |
|---|---|---|
| 50 | 1.000 | 1.000 |
| 100 | 0.999 | 0.999 |
| 150 | 0.998 | 0.998 |
| 200 | 0.994 | 0.994 |
| 250 | 0.991 | 0.992 |
| 300 | 0.982 | 0.983 |
| 350 | 0.976 | 0.977 |
| 400 | 0.960 | 0.961 |
| 450 | 0.936 | 0.937 |
| 500 | 0.870 | 0.871 |
| 550 | 0.559 | 0.562 |
| 600 | 0.400 | 0.409 |
| 650 | 0.036 | 0.036 |
| 700 | 0.031 | 0.032 |
| 750 | 0.029 | 0.029 |
These data indicate that length-based filtering isn't going to be a particularly thorny issue, which is expected given that BOLD as a database is interested in acquiring most data in the 500-650 bp regions for submission acceptance. Nevertheless, I wasn't sure at all about how homopolymer-based filtering would work, and what those expectations might be. While RESCRIPt has a function to perform this kind of filtering, the parameter is a single integer applied to all bases equally. Given that different regions of COI might have different homopolymer variation, and that the database itself might consist of different sequencing platforms that are more or less suceptible to creating homopolymer data, I tallied the number of homopolymers in every sequence in the degapped dataset, for homopolymer lengths between 5-15 bp, for each character. This required creating a fasta file, homopolymer_motifs.fasta that looked a bit like this:
>seq1
AAAAA
>seq2
AAAAAA
>seq3
AAAAAAA
...
>seq42
TTTTTTTTTTTTT
>seq43
TTTTTTTTTTTTTT
>seq44
TTTTTTTTTTTTTTT
I used a package, seqkit, which makes searching the patterns in the fasta file easy:
seqkit locate -P -f homopolymer_motifs.fasta ./degap_tmpdir/dna-sequences.fasta --threads 4 | \
awk -v OFS="\t" 'NR>1 {print $1, $3}' | awk '{print $1, length($2)}' | \
sort | uniq | awk '{print $3}' | sort | uniq -c > homopolymer_motifs.table
importantly, we're not double counting instances where a sequence may contain multiple homopolymers of different DNA bases with the same length - we're collapsing that information and counting the number of sequences with one or more homopolymers of any non-ambiguous DNA base character
The homopolymer_motifs.table file is then used to generate the plot below. We see that using a homopolymer value of 8 or less would result in losing the majority of our sequences - let's don't do that! Instead, a value of 9 or greater likely would suffice to remove sequences that may indicate spurious sequences. Note that the values above the bar indicate the number of sequences with at least 1 homopolymer - for example, there are 89 sequences that have at least one A, T, C, or G base that is repeated 15 consecutive times!
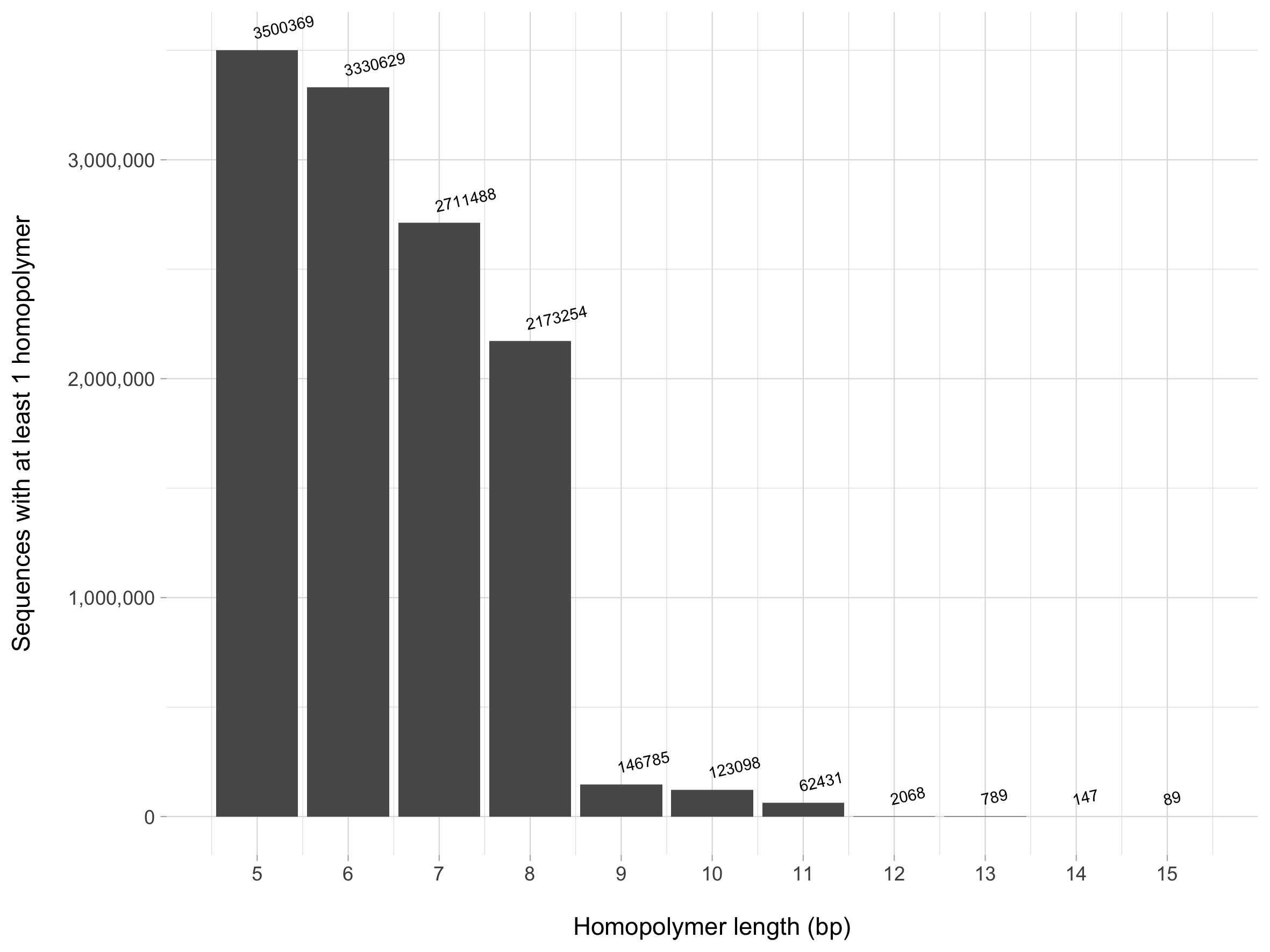
So where does this leave us in creating the database? There are many tasks we still need to address, some of which I think are broadly applicable to every user regardless of their end goal, some of which are user-specific:
General
- Removing sequences outside of some length threshold (lower and/or higher!)
- Removing trailing/leading ambiguous nucleotides.
- Removing sequences with large numbers of "internal" ambiguous nucleotides.
- Removing sequences with large stretches of homopolymers.
User specific - Dereplicating the data.
- Retaining only those sequences that are applicable to the COI region we have amplified.
We might be better off starting at the end, and tackling point 6 about selecting only the portions of these raw data that align to your primers. If you do that first, it's quite possible that a lot of the length, ambiguous nucleotide, and homopolymer quality-control filtering might ultimately not apply to the parts of the COI amplicon you aren't even concerned with (as a lot of these issues arise at the ends of fragments). Nevertheless, restricting COI regions by primer can get tricky: a wise rotifer biologist once pointed out to me that it's possible to have a sequence in a database that was amplified by the same primers you're trimming with, but before that reference sequence was submitted, the researcher trimmed off the primers - see this post for more details. As a result, you can lose otherwise good data by requiring particular primer regions! There's also the issue of how specfic a database should be, and whether or not a single primer set is useful for your own research. Perhaps it would be more sensible to expand on that one primer boundary and include a handful of other primers as well that other researchers might use in neighboring regions. In my work with arthropod COI data, for example, a series of alternative forward and reverse primers exist, all of which might be used by different researchers to amplify different COI regions.
In fact, that same database wizard biologist offered a totally different way of thinking about parsing these data, such that rather than trimming by primer sequence, we figure out where the regions of interest are based on alignment. This takes longer, but should retain those instances where our reference sequences are missing the primer bits, but retain everything else. This work was described in full detail for processing a SILVA dataset outside of RESCRIPt - in partiuclar, we're stealing all the good stuff from step 7.
Unfortunately for us, unlike that SILVA workflow, we don't have any alignment file to start with, so we're going to throw some serious computational power to get this to work. We'll take the following approach:
- Remove any degapped sequences with more than 5 ambiguous nucleotides or at least 12 homopolymer characters. Notably I'm starting with the degapped fasta file that was exported from the
bold_degapSeqs_min1.qzafile. RESCRIPt developers will likely have a parameter to do this work directly on the file in the near future. - Of the remaining sequences, remove any shorter than 250 bp and longer than 1600 bp.
- Dereplicate these data.
- Perform a massive alignment of all of the remaining, dereplicated sequences.
- Use a particular primer pair to identify the boundaries that we want to retain among our COI sequences. Then use these boundary coordinates to trim our dataset to those regions.
- Remove gaps again from the previously aligned (and now Folmer-region-specific) sequences, and filter for length (250bp or less are removed).
- Dereplicate a second time (because we care about just this region now)!
If that all works, we should have a nice COI database ready for classification (as well as ready for further evaluation within RESCRIPt!).
Steps 1-2: Quality control filtering
The first step is to create a fasta file with prefix/suffix ambiguous bases removed. At the moment, I had to do this manually outside of QIIME (but stay tuned for potential parameters to do this within qiime rescript degap-seqs!). I then import that file back as a .qza object; because we no longer have gaps in our data, we change the --type too.
Recall that the file
dna-sequences.fastawas obtained by exporting thebold_degapSeqs_min1.qzaobject earlier.
cat ./degap_tmpdir/dna-sequences.fasta | paste - - | \
awk -v OFS="\t" '{gsub(/^[N]+/,"",$2);print}' | \ ## removes prefix N's
rev | sed 's/^N*//' | \ ## removes suffix N's
rev | tr '\t' '\n' > bold_outerNtrimmed.fasta
qiime tools import \
--input-path bold_outerNtrimmed.fasta \
--output-path bold_outerNtrimmed.qza \
--type 'FeatureData[Sequence]'
We can now accomplish the first two step to (1) remove sequences with excessive numbers of ambiguous nucleotides and/or homopolymers, and (2) filter by length. Both of these are accomplished by a pair of RESCRIPt commands:
qiime rescript cull-seqs \
--i-sequences bold_outerNtrimmed.qza \
--p-num-degenerates 5 \
--p-homopolymer-length 12 \
--o-clean-sequences bold_ambi_hpoly_filtd_seqs.qza
qiime rescript filter-seqs-length \
--i-sequences bold_ambi_hpoly_filtd_seqs.qza \
--p-global-min 250 \
--p-global-max 1600 \
--o-filtered-seqs bold_ambi_hpoly_length_filtd_seqs.qza \
--o-discarded-seqs bold_ambi_hpoly_length_discarded_seqs.qza
There are now 3,464,989 total sequences
The bold_ambi_hpoly_length_filtd_seqs.qza object has sequences filtered for ambiguous nucleotide content, homopolymers, and a minimum and maximum length. We're getting somewhere!
Step 3 - Dereplicating (part 1)
We're dereplicating these data using a RESCRIPt function that is called a "super" mode. In brief, we are applying a Least Common Ancestor (LCA) method, while giving preference to those sequences that are represented more often than another. For example, say we had 4 identical sequences in our original dataset, with the following taxonomies:
>100;tax=k__Animalia;p__Annelida;c__Polychaeta;o__Phyllodocida;f__Nereididae;g__;s__
>101;tax=k__Animalia;p__Annelida;c__Polychaeta;o__Phyllodocida;f__Nereididae;g__Neanthes;s__Neanthes fucata
>102;tax=k__Animalia;p__Annelida;c__Polychaeta;o__Phyllodocida;f__Nereididae;g__Neanthes;s__Neanthes fucata
>103;tax=k__Animalia;p__Annelida;c__Polychaeta;o__Phyllodocida;f__Nereididae;g__Neanthes;s__Neanthes fucata
What would happen in this case is we would retain the full taxonomic description including the species-rank information because the majority of the sequences contain those identical labels. The assumption we're making is that the first sequence (>100) was an incomplete description. If we didn't prefer the more frequent label, then the final record would lose genus and species rank information. It's also true that if the majority of sequences lack information at a given rank, then even those other identical sequences with that information are discarded.
For instance, in the following scenario:
>201;tax=k__Animalia;p__Arthropoda;c__Insecta;o__Coleoptera;f__Scarabaeidae;g__Serica;s__
>202;tax=k__Animalia;p__Arthropoda;c__Insecta;o__Coleoptera;f__Scarabaeidae;g__Serica;s__
>203;tax=k__Animalia;p__Arthropoda;c__Insecta;o__Coleoptera;f__Scarabaeidae;g__Serica;s__
>204;tax=k__Animalia;p__Arthropoda;c__Insecta;o__Coleoptera;f__Scarabaeidae;g__Serica;s__Serica porcula
we would discard the species information and retain a record that contained only the genus name.
On to dereplicating these data. This should cut our dataset down by at least half, easing the burden on the subsequent multiple sequence alignment process. The output sequence object, dereplicated-sequences bold_derep1_seqs.qza is exported and that fasta file is then used to perform the multiple alignment step. Note that the --p-derep-prefix flag is another trick to reduce redundancy: in this case, if one sequence is an exact subset of another sequence, we keep the longer of the two sequences. FWIW, when I didn't use this flag the first time, I retained about 500,000 additional sequences than the second time I ran the script with it. When you have to do a pairwise alignment of every sequence, every little bit counts (squared).
qiime rescript dereplicate \
--i-sequences bold_ambi_hpoly_length_filtd_seqs.qza \
--i-taxa bold_rawTaxa.qza \
--p-mode 'super' \
--p-derep-prefix \
--p-rank-handles 'greengenes' \
--o-dereplicated-sequences bold_derep1_seqs.qza \
--o-dereplicated-taxa bold_derep1_taxa.qza
Dereplcating these data reduced the number of sequences from 3,464,989 to 1,718,762.
minor point: adding the
--p-derep-prefixflag makes a substantial difference. I ran the same command without it and we retain over 2.2 million sequences instead of 1.7 million.
One teeny thing I noticed only after going through this workflow a few times and getting errors in downstream evaluations: you may want to perform an additional filter of those dereplicated sequences so that you don't retain sequences that contain very little taxonomic information. For example, if you into the taxonomy strings associated with the bold_derep1_taxa.qza object, you'll find that there are 32 sequences have this taxonomic info:
tax=k__Animalia;p__;c__;o__;f__;g__;s__
notably, the only data missing Phylum-rank information are within the Animal domain/kingdom
Depending on your own database inputs, it might be the case that there is a fair bit of missing taxonomic information remaining. In our case, it's relatively minimal, but as an example, we can run the following code to remove those sequences that lack Phylum-rank information. Note that I didn't actually apply this filter - I'm just showing it so that you have a sense of what actions to take if needed.
## filter taxonomy file
qiime rescript filter-taxa \
--i-taxonomy bold_derep1_taxa.qza \
--p-exclude 'p__;' \
--o-filtered-taxonomy bold_derep1_PhylumFiltd_taxa.qza
## filter sequences file
qiime taxa filter-seqs \
--i-sequences bold_derep1_seqs.qza \
--i-taxonomy bold_derep1_taxa.qza \
--p-exclude 'p__;' \
--o-filtered-sequences bold_derep1_PhylumFiltd_seqs.qza
Step 4 - (Multiple!) multiple sequence alignments
My goal was to create a COI database that was bounded by a particular primer sequence. For instance, in my animal diet work, our lab uses ANML primers, which create amplicons of around 180 bp. There are lots of other COI primer sets out there, and most are derivations to amplify a sequence bound within one of the original primer sets for COI work (a.k.a. the Folmer primers). The goal here is to identify the coordinates where the primers align to the reference sequences, then remove any up and downstream sequence bits outside of the primer pair of interest. In this example, we'll use the ANML primer pair, but you can use whatever pair of primers you want!
The process consists of a series of multiple sequence alignment (MSA) steps, beginning with the dereplicated sequences created from the previous step - bold_derep1_seqs.qza. The first step requires aligning a subset of high quality sequences to themselves. Next, the primers of interest are added to this subsetted reference alignment. Finally, we create a giant alignment by adding in all the remaining sequences from our original reference set (except those seqs that were subset out to create the initial small alignment). When all that alignment work is done, we'll move on to removing all of the up/downstream sequences, remove the gaps in the aligned sequences, and generage a reference dataset with only sequences information specific to the primers we care about. Nice!
Step 4a: Aligning a subset of sequences from our giant reference dataset
This first part is critical, and I struggled to get this right the first half dozen tries. The goal is to first identify a subset of high quality sequences that will align to each other without producing a huge number of gaps. Given that the majority of COI sequences are from insects, and are usually around 658 bp, I started by exporting the bold_derep1_*.qza files from the previous step, then filtering those dereplicated fasta sequences. The options below can certainly be achieved in QIIME, but I leaned heavily on the seqkit package to perform these operations:
First, export the bold_derep1_seqs.qza and bold_derep1_taxa.qza objects:
qiime tools export --input-path bold_derep1_seqs.qza --output-path tmpdir_boldFullSeqs
qiime tools export --input-path bold_derep1_taxa.qza --output-path tmpdir_boldFullTaxa
Next, filter for min/max length:
seqkit seq --min-len 660 --max-len 1000 -w 0 ./tmpdir_boldFullSeqs/dna-sequences.fasta > boldFull_lengthFiltd_seqs.fasta
grep -c '^>' boldFull_lengthFiltd_seqs.fasta
669,615 seqs remain
Now we will search back through our taxonomy file to identify only those FeatureIDs labeled as insects and contain complete species descriptions. We then select just those length-filtered sequences from the insect.list file:
## generate the .list
grep 'p__Arthropoda;c__Insecta' ./tmpdir_boldFullTaxa/taxonomy.tsv | \
grep -v 's__$' | grep -v 's__.*sp.$' | grep -v '-' | cut -f 1 > insect_species.list
## filter the sequences to include only those in the .list
seqkit grep --pattern-file insect_species.list boldFull_lengthFiltd_seqs.fasta > boldFull_lengthNtaxaFiltd_seqs.fasta
grep -c '^>' boldFull_lengthNtaxaFiltd_seqs.fasta
There are 510,105 in the insect_species.list file; 246,650 of these pass the length filter from above (246,650 seqs retained).
We'll perform one final filter where we retain only sequences with non-ambiguous bases (must be only A|C|G|T):
seqkit fx2tab -n -i -a boldFull_lengthNtaxaFiltd_seqs.fasta | awk '{if ($2 == "ACGT") print $1}' > nonAmbig_featureIDs.txt
seqkit grep --pattern-file nonAmbig_featureIDs.txt boldFull_lengthNtaxaFiltd_seqs.fasta -w 0 > boldFull_lengthNtaxaNambigFiltd_seqs.fasta
grep -c '^>' boldFull_lengthNtaxaNambigFiltd_seqs.fasta
228,351 seqs remain
We'll subsample those filtered and aligned sequences to just the first 2,000:
seqkit sample --rand-seed 101 --number 2000 --two-pass boldFull_lengthNtaxaNambigFiltd_seqs.fasta -w 0 > bold_mafftRefs_subseqs_seqs.fasta
CAUTION: Heads up on that last step!! This was the part that really threw me for a loop, and likely takes some trial and error depending on your primer set and reference sequences. It may be that subsetting too many or too few random sequences incorporates too much variability in your alignment, and messes with the subsequent mapping of primers to the MSA file (you're about to make in the next step). For example, when I subsetted just 1,000 sequences (instead of 10,000) I noticed that my primers wouldn't align to the MSA file properly. When I subsetted to 10,000 sequences, it gave a different set of primer mapping coordinates that were also incorrect (more on this explained in the next section). The sweet spot I found for this subsample was 2,000 sequences. But it might have nothing to do with the particular number of random sequences you collect either - it might be that including sequences of certain lengths are useful or problematic. Or it could be because of the sequence diversity among the group (maybe don't try to align cnidarians with hemipterans in your few samples, for instance). The general point I'd raise here is that if you struggle to get your primers to align properly, consider any or all of these filtering steps above. Maybe change the taxonomic representation to be more refined (maybe just do it with a single Family or Genus, for example). Maybe change the length of sequences. The goal is always going to be to find somewhere over (at least) 100 high quality reference sequences that you trust will (1) align with few gaps, and (2) contain both the primer sequences you're going to align. Unfortunately, there's no magic trick here. Just trial, error, coffee, more error, and a bunch of silent (or vocal) cursing.
We'll create an initial alignment of just these 2,000 reference sequences using MAFFT (I installed v7.471). Also note that it's worth your while to specify the temporary directory where mafft will put all the files. It won't matter for just these first thousand sequences, but it's possible that you can max out the default /tmp folder when you're running bigger alignments in later steps. I just specify it up front here so I don't forget it later ![]()
The process should be really fast - expect to complete the alignment in under a minute if you have more than 12 CPUs at your disposal (though this obviously will vary on the length and number of sequences in your own alignment):
export MAFFT_TMPDIR=$(pwd) ## change this path to suit your needs
mafft --auto --thread -1 bold_mafftRefs_subseqs_seqs.fasta > reference_MSA
note the output here mentions the method used by
--auto:
Strategy:
FFT-NS-2 (Fast but rough)
Progressive method (guide trees were built 2 times.)
We can now use that reference_MSA to guide our next steps.
Step 4b: aligning the small reference set to the primers of interest
We're going to use the ANML primers described above. We'll use the standard 5'->3' orientation for the forward primer, but take the reverse complement of the listed 5'->3' sequence for the reverse primer. Thus, whatever primer you end up using, just make sure to take the reverse complement of the reverse primer's 5'-->3' orientation (if you're really unsure, just add a bunch of possible forward/reverse/reversecomplement orientations to your fasta file and see what aligns!). These primer sequences are put into a fasta file (let's call it anml_primers.fasta) formatted like this:
>PRIMER_ANML-f
ggtcaacaaatcataaagatattgg
>PRIMER_ANML-r
ggatttggaaattgattagtwcc
Now we'll align these primer sequences to our small alignment created in the last step. This step is also very fast, again assuming you have just a pair of primers aligning to around the same number (2000) of sequences as I am aligning. Expect under a minute to complete:
mafft --multipair --addfragments anml_primers.fasta --keeplength --thread -1 --mapout --reorder reference_MSA > ref_primer
the
--mapoutflag creates an output file,anml_primers.fasta.map, which contains information regarding the coordinate positions of where our primers are located within the small alignment. These values should be sufficient for our subsequent filtering of up/downstream sequences once the giant alignment is created, but we'll run this same option later to double check that the primer coordinate positions haven't changed once the larger alignment is built.
This step is a little strange when you realize that we're going to be essentially duplicating the actions again in a few steps. Why do things twice? It's really a sanity check to confirm that the primers you've selected are actually going to map to the reference sequences you've retained. It's quick to get this info for the small dataset (and takes a lot more effort to do with the giant alignment). Thus, we're aligning our primers against this small set of references to confirm they are indeed mapping in the right location by viewing the contents of that anml_primers.fasta.map file. What do we see in that file? It contains information regarding the primer sequence character (column one), the position of that sequence character internal to the primer sequence itserlf (column two), and the position of that sequence character relative to the alignment file (third column). It looks like this (I've added the ... to shortern up the view to include just the first and last pair of nucleotides in the primer alignment):
>PRIMER_ANML-f:
# letter, position in the original sequence, position in the reference alignment
g, 1, 17
g, 2, 18
...
g, 24, 40
g, 25, 41
>PRIMER_ANML-r:
# letter, position in the original sequence, position in the reference alignment
g, 1, 249
g, 2, 250
...
c, 22, 300
c, 23, 301
In other words, our forward primer, PRIMER_ANML-f begins at coordinate position 17 of the ref_primer alignment, and ends at position 41. This primer is 25 bases long (the second column starts at 1 and ends at 25), so in this particular case there no gaps in the alignment within the forward primer (41 - 17 + 1). Our reverse primer, PRIMER_ANML-r, begins at position 249 of the alignment file, and ends at position 301. This primer is only 23 bases long, yet there are 53 positions spanning the length of the primer - why? alignment gaps.
I relied on a Python script, extract_alignment_region.py, to check out what would happened if we trimmed our alignment at those primer flanks - would the resulting fragments, once gaps are removed, be the expected length of our amplicon when the specific primers are used? In the script below, we're going to enter coordinate positions for the valueStart and valueEnd positions based on the information in the anml_primers.fasta.map file. Specifically, we want to valueStart to be one base longer than the largest value of the forward left flank coordinate, and the valueEnd to be one base shorter than the smallest value on the right flank coordiante. In our case, that would be 42 for >PRIMER_ANML-r and 248 for >PRIMER_ANML-f:.
The following script is courtesy of a generous rotiferphile (if that's a thing?), Mike Robeson.
python extract_alignment_region.py \
-i ref_primer \
-o ref_primer_coordinateTrimmed.fasta \
-s 42 \
-e 248
We then use that fasta file, ref_primer_coordinateTrimmed.fasta, remove any remaining gaps, and cound the lengths of the remaining sequences. If we aligned at the right space, the majority of these sequences should be around 180 bp - the length of the amplicons we generate with the ANML primers, as shown in this COI primer map - when you look at the labels on that map, we're using the LepF1-LCO1490 as ANMLf, and CO1-CFMRa, as ANMLr.
seqkit seq -g -w0 ref_primer_coordinateTrimmed.fasta | paste - - | awk '{print length ($2)}' | sort | uniq -c
What we find is that indeed, the vast majority of our sequences are exactly 181 bp (1714 of 2000 samples), and about 97% of these subsambled seqs are between 177-181 bp (1939 of 2000), so we're good to go! Unfortunately, if you don't get this right, your entire database will be trimmed to a primer coordinate that you aren't expecting. Once you get things aligned to the expected positions and of the expected lengths, you can move on to the giant alignment step. Let's tackle that next!
Step 4c: aligning all the rest of the sequences
Now for the much heavier task: aligning the million+ references to this comparatively tiny alignment of just 2,002 sequences (our initial 2,000 reference sequences plus our two primers). Let's start by filtering the complete reference artifact, bold_derep1_seqs.qza, to remove the 2,000 reference sequences already aligned, then exporting that file into a traditional fasta format:
## create a list of the sequences to drop:
grep '^>' bold_mafftRefs_subseqs_seqs.fasta | sed 's/^>//' > droplist.txt
## filter OUT the sequences to include only those NOT in the droplist.txt file
seqkit grep -v --pattern-file droplist.txt ./tmpdir_boldFull/dna-sequences.fasta > seqsForGiantAlignment.fasta
This subsetted
seqsForGiantAlignment.fastafasta contains 1,716,762 sequences - exactly 2,000 fewer than we started. That's what we expect, given that we used 2,000 of these to generate our initialreference_MSAfile. We'll now add these 1.7 million sequences to generate a giant alignment!
At long last - to the giant alignment! We align the remaining reference sequences to our small alignment file, ref_primer as follows:
export MAFFT_TMPDIR=$(pwd) ## change this path to suit your needs (in case you forgot the first time!)
mafft --auto --addfull seqsForGiantAlignment.fasta --keeplength --thread -1 ref_primer > giant_alignment
This step takes a large amount of memory to run. I ended up requiring around 150 GB RAM to get it to complete. The job finished in about 60 minutes using 24 CPUs (I've run the same task on another machine with 8 CPUs and it took about 3 hours to finish, so it seems to scale pretty linearly on the number of threads available). The mafft alignment strategy used was: FFT-NS-full
The output, giant_alignment, contains the information we need to now: (1) identify the alignment positions where our primers are bound, and then (2) trim all sequences up and downstream from those positions.
Step 5 - Using the alignment information to create a primer-bound reference dataset
We'll use the MAFFT output giant_alignment file in the next step in two ways: (A) We'll perform a sanity check by aligning those same primers to a subset of sequences from the giant file and gather the coordinates where they are bound. Recall back in step #4a that we created an output file that contained the coordinate positions of the primers relative to the small alignment - we want to double check that this new alignment has primers aligning to the same positions. It doesn't matter what the subset of sequences are, because the alignment length for all sequences is fixed. Then (B), we apply a Python script to trim the aligned bold_derep1_mafft.out file based on those coordinate boundaries (rather than the primers themselves!).
Step 5a - Confirmation of primer coordinate positions in giant alignment file
Rename the original output map file, to a new name to avoid overwriting in the next step:
mv anml_primers.fasta.map anml_primers.fasta.map_toSmall
Subset the giant alignment file to retain just the first 2,000 sequences. Forutnately, the seqkit package is savvy enough to deal with line-wrapped fastas (i.e. it doesn't matter if the fasta sequence is on one line or many - you'll get 2,000 unique sequences this way):
seqkit head -n 2000 giant_alignment > subset_giant_alignment
Align those same primer sequences used earlier to the subsetted giant alignment file, subset_giant_alignment. Note we don't actually need to retain the output fasta file copy_giant_alignment_wdupPrimers - we just want to confirm the coordinate positions of the primer sequences!
mafft --multipair --addfragments anml_primers.fasta --keeplength --thread -1 --mapout subset_giant_alignment > subset_giant_alignment_secondaryPrimerAlign
We can ignore the subset_giant_alignment_secondaryPrimerAlign output. The information we want is in the Let's rename the anml_primers.fasta output file. Let's rename this anml_primers.fasta output to keep consistent with the naming convention used earlier too when we renamed the original .map file:
mv anml_primers.fasta.map anml_primers.fasta.map_toGiant
What we want to investigate are the coordinate positions of those primers in the anml_primers.fasta.map_toGiant file:
>PRIMER_ANML-f:
# letter, position in the original sequence, position in the reference alignment
g, 1, 17
g, 2, 18
...
g, 24, 40
g, 25, 41
>PRIMER_ANML-r:
# letter, position in the original sequence, position in the reference alignment
g, 1, 249
g, 2, 250
...
c, 22, 300
c, 23, 301
Sure enought, nothing has changed - perfect! That's what we expected given that we passed the --keeplength flags in our mafft alignments, but it's always good to be sure.
With our primers confirmed to be aligned to the right coordinates in our large alignment file, we can now move to the final steps of this part of the workflow: trimming the any up/downstream bases outside of our primer coordinates.
Step 5b - trimming the giant alignment to include only sequences within our primer coordinates
With the ANML-primer boundaries defined, the giant alignment of all our reference sequences, giant_alignment, can be sliced such that all of our database contains sequences in just the COI region our amplicon data is expected to align to. Once again, the heavy lifting is done via the Python script used earlier, extract_alignment_region.py. We'll use the same coordinate positions for the valueStart and valueEnd positions as before:
python extract_alignment_region.py \
-i giant_alignment \
-o bold_derep1_anml_tmp.fasta \
-s 42 \
-e 248
and volia! we have a trimmed dataset with a defined boudary without having to require any primers be detected within the reference sequences themselves. One last thing before we wrap up this section - remember how we've added the primer sequences into this alignment file? We obviously don't want to keep them in our final dataset (they aren't reference sequences, after all!). The neat thing here is that if we've trimmed the correct positions, those primers themselves should be cut out. Let's double check on this:
grep PRIMER --color -A 1 bold_derep1_anml_tmp.fasta
You'll notice the output looks something like this:
>PRIMER_ANML-r
---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
--
>PRIMER_ANML-f
---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
... just a bunch of gaps - no primers left. Looks like our coordinate trimming worked as expected!
The last thing to do is remove those two primer sequences to have our final alignment file. We'll use seqkit once more to make this fast and simple:
## make a list of sequence headers to drop:
grep PRIMER bold_derep1_anml_tmp.fasta | sed 's/>//' | sed 's/[ \t]*$//' > dropPrimers.txt
## exclude these sequences from the alignment file:
seqkit grep -v -f dropPrimers.txt bold_derep1_anml_tmp.fasta | seqkit seq --upper-case -w 0 > bold_derep1_anml.fasta
There are 1,718,762 sequences in
bold_derep1_anml.fasta- good. That's the number of sequences we started with when we were aligning the sequences from thebold_derep1_seqs.qzafile!
Step 6 - Removing gaps, filtering by length
We're almost finished with this database design (I promise)! Next up is to remove the gaps of this aligned file, then filter the sequences by length. I was curious how the alignment strategy above would impact the sequence lengths of the remaining fragments. After all, not every sequence in our reference dataset would necessarily contain sequence spanning the entire ANML primer region - some might in fact contain none of that region, or just a part. I thus calculated the length of each sequence, and came up with this histogram representing the frequency of sequence lengths of the x file.
The calculation was...
grep -v '^>' bold_derep1_anml.fasta | sed 's/-//g' | awk '{print length}' | sort | uniq -c > anml_seq_lengths.freq.table
... and the resulting plot of that anml_seq_lengths.freq.table file, created with this R script, looks like this:
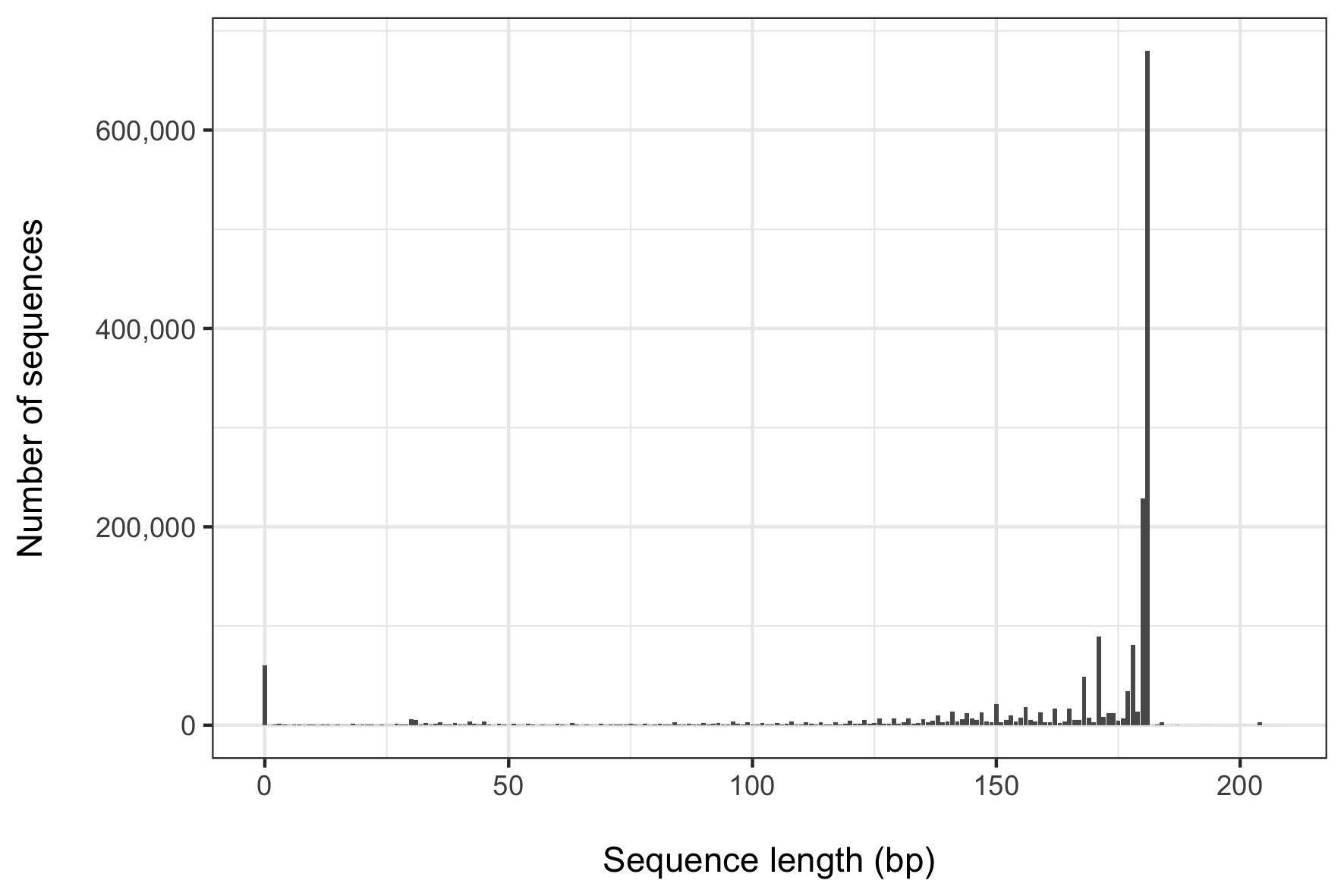
What becomes apparent when visualizing these results is that the majority (~ 53%) of our sequences are represented by just two lengths (180 and 181 bp), and about 80% of sequences are represented by fragments between 150 to 180 bp. It's also worth noting that there were 60,664 sequences with a length of zero. Clearly we want to filter those out! I found that visualizing these data was helpful to inform me about what sequence length would be appropriate for the following command. I chose 170 bp to try to balance the desire to retain as many distinct sequences as possible while wanting to retain only as near full-length a sequence as possible. At 170 bp, I still retain about 69% of all sequences (over 1.25 million!).
Start by importing this file back into QIIME (as all remaining tasks will occur in that environment). Then remove gaps and filter sequences by length:
qiime tools import \
--input-path bold_derep1_anml.fasta \
--output-path bold_derep1_anml_seqs_aligned.qza \
--type 'FeatureData[AlignedSequence]'
qiime rescript degap-seqs \
--i-aligned-sequences bold_derep1_anml_seqs_aligned.qza \
--p-min-length 170 \
--o-degapped-sequences bold_derep1_anml_seqs_nogaps.qza
Following the length-filtering of the gap-removed sequences, we have 1,182,895 reference COI sequences remaining
We now have one step to go - using the bold_derep1_anml_seqs_nogaps.qza file as input for a second dereplication step.
Step 7 - Dereplicating (part 2)
Wait - didn't we do this already? The idea behind a second dereplication strategy is that it's possible that two sequences might have identical sequences within our now ANML-primer-trimmed boundary, but had initially different sequence composition outside of that boundary. For example, you can imagine that an initial pair of sequences looked like:
| <---- ANML region -----> | <--- trimmed region ---> |
seq1: AATTGGCCAATTCCGGAATTCCGGCCGGAAAATTTTCCCCGGGGATATATATCGC
seq2: AATTGGCCAATTCCGGAATTCCGGCCGGAAGGGGGGGAAAAAAAACCCCCCTTTT
The two sequences are clearly different across their full lengths, but once we were to slice only those regions within the primer region, there's a chance that we now have duplicate sequences. It's possible you want to keep these duplicate sequences if they represent different taxonomies - see the subsection in the dereplication of sequences and taxonomy portion of the SILVA RESCRIPt tutorial on "Dereplication considerations" for commentary on why you might do this. In my case, I'm opting for a single consensus taxonomy for each unique sequence, so I am going to dereplicate using the same strategy as before:
qiime rescript dereplicate \
--i-sequences bold_derep1_anml_seqs_nogaps.qza \
--i-taxa bold_rawTaxa.qza \
--p-mode 'super' \
--p-derep-prefix \
--p-rank-handles 'greengenes' \
--o-dereplicated-sequences bold_anml_seqs.qza \
--o-dereplicated-taxa bold_anml_taxa.qza
Once this dereplication step is complete, we have 739,345 unique COI sequences. That's it, we're at the end of the database construction section - we made it! Considering that we started with over 3 million COI sequences from BOLD, clearly all these filtering steps have made an impact.
The road(s) ahead
What work remains to be done?
-
While this database workflow accessed only BOLD references, it's quite likely that incorporating other references from alterantive resources would improve our classifiers. Shameless plug: I explored just this question in a recent paper. Fortunately, the RESCRIPt crew has developed a tool,
get-ncbi-datato pull NCBI GenBank data right into a QIIME formatted database. It's my intention on expanding this tutorial to show how we might include both BOLD and NCBI references into an improved COI database. -
Obviously, we haven't even touched on any of the evaluation steps available within RESCRIPt! I'm still working on these tests at the moment and will update this document to reflect those findings. Fortunately, all the tests rely specifically on RESCRIPt commands - provided you have a database ready to go, just follow the evaluation functions in the RESCRIPt tutorial. Critically, one of these functions allows for an evaluation of a naive Bayes classifier, and produces a classifier file in the process! Thus far, I've created just a single classifier for the
bold_anmldataset, but plan on creating on for thebold_derep1dataset also. Please check back for updates on this progress.
The classifier was created as follows:
qiime rescript evaluate-fit-classifier \
--i-sequences bold_anml_seqs.qza \
--i-taxonomy bold_anml_taxa.qza \
--p-reads-per-batch 6000 \
--p-n-jobs 6 \
--output-dir fitClassifier_boldANML
This was the most memory-intesive process of the entire workflow! The job completed in about three days using approximately 140 GB RAM
The bold_anml_classifier.qza file is available here
![]() !! additional file Update!!
!! additional file Update!! ![]()
I recently created a classifier file using the untrimmed COI sequences for Arthropod taxa only - this is saved as bold_full_ArthOnly_classifier.qza, and I've hosted it here. Just a head's up - it's a big file (~500 Mb)!