Dear qiime-support,
I have sequences from a 2 x 250 bp Illumina run. The desired product is 394 bp long, therefore I guess there should be enough overlap in the merging step. I tried several possibilities of truncation (2 x220 bp, 2x 215), e.g.
The problem is, that I get to less reads after merging, this should not be the problem after quality check. Or have I missed something?
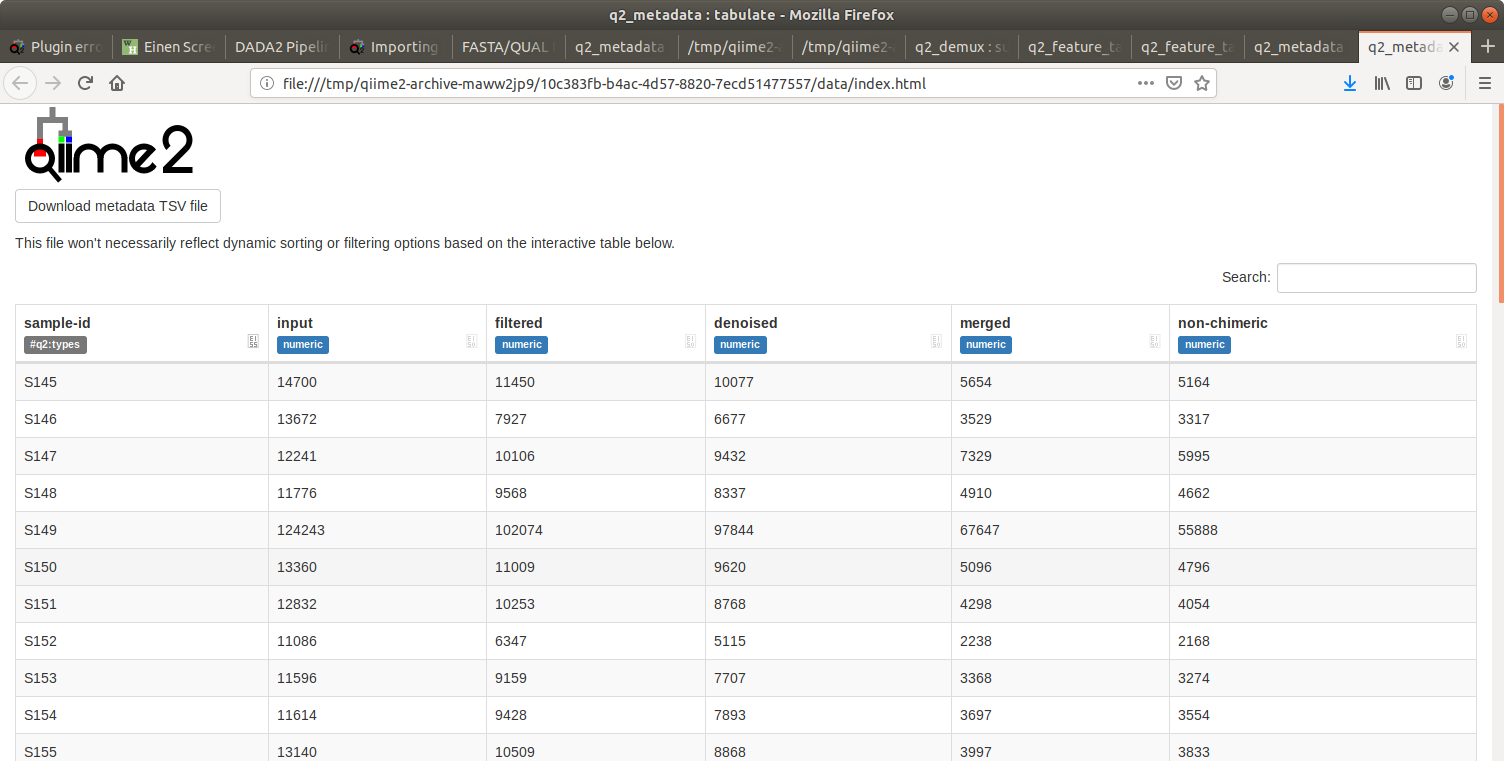
I have made some screenshots of the quality after primer trimming and the table after denoising!
We use qiime2-2019.7 (conda).
Reading the other posts, it might still be a possibility that the overlap is to small, but I don't see why. Merging with Usearch resulted over 92 % merged sequences.
Given the information you provided, you should be getting more successful merges. Quality looks good and the reads are long!
So we need a little more information:
What is the target?
Is that 394 bp before or after primers have been trimmed?
Are the primers already trimmed from these reads?
How much length variation do you anticipate in your amplicon?
What trimming parameters did you use prior to merging with usearch? If they were different, the reason is clear and you should try to replicate those trimming parameters here. Usearch may also require a different minimum overlap from dada2 (which now requires ≥ 12 nt overlap).
But while you answer those, maybe try truncating at 230 x 230 just to see what happens.
By the way, instead of posting screenshots to the forum you can upload your QZV files instead (if they don't contain sensitive information, which dada2 stats and quality score plots shouldn't really) — then we can view the plots directly here, which can often be helpful for diagnosing issues.
Dear Nicolas,
thank you for your rapid response,
I have a 16S primer system 799f-1193r (used to discriminate between plastids and bacterial DNA) targeting the V5-V7 region. The quality picture I sent you was after trimming, so that I used trimmed sequences for the denoising step (I have checked again).
For the merging with usearch I used the standard approach (usearch -fastq_mergepairs fastqout merged_1.fq -tabbedout merged.txt , therefore default setting max-diff = 5).
I tried the truncation with 2 x 230, but in this case, nearly all sequences are filtered out: denoising-stats2.qzv (1.2 MB).
If I go down to 2 x 220 I get more sequences out: denoising-stats.qzv (1.2 MB)
Hi @kfri,
I am not sure what is going wrong — with everything you describe it sounds like you should get sufficient overlap but you are still losing ~50% of your reads at merging. My guess is that there is more variation in the amplicon length than you may expect — I do not know enough about that primer pair, but you could use extract-reads to estimate amplicon size as described here:
But that does not explain why usearch merge_pairs is giving you better merging; I suspect it could be because dada2 is trimming just a little bit prior to the merge, enough to miss the mark for half your sequences. You could use qiime vsearch join-pairs to join the reads first, then use q2-deblur for denoising. join-pairs would be similar to usearch fastq_mergepairs so we should see similar results — maybe give that a try and see if you still see sufficient merging?
Hi Nicolas,
I used the vsearch join-pairs command, and get similar results (up to 90 % merging, view joint587-filtered.qzv (306.7 KB) ), but if I run these sequences with the deblur algorithm (I used the 16S version, but is this for the full-length 16S? Unfortunately I haven't found the information.), I also get low sequence amounts.
I also started to run an old (good working) dataset, but this looks quite the same. I guess I will go on trying, but maybe for this data I have to go back to OTU clustering.
Thanks a lot for your help!
deblur denoise-16S is for any region of 16S. The idea is that it performs a rough "positive filter" to make sure the input sequences resemble any portion of 16S.
Could you please post the deblur denoising stats? These could be very informative for diagnosing why these sequences are failing to pass deblur.
I would discourage that... the fact that these are failing with deblur tells me that they probably have something wrong with them that OTU clustering would not catch. This is part of the motivation behind deblur — OTU clustering just clusters, but does not denoise and hence can yield large amounts of junk! This may be one of those scenarios, e.g., where there is a large amount of non-target DNA that is being filtered by deblur.
I had a look over old runs and old merging results and I guess it might be that the matching is not long enough, because parts of the community seem to have longer 16S. These sequences are deleted by the truncation. If I use vsearch the joined sequences are something maximum 385 bp, but if I look in the old pipeline the sequences were something over 400 bp after all trimming. I guess this is the problem and I am afraid that due to the trimming, I would change the microbial community distribution (due to trimming away 16S sequences which are longer). joint587-filtered.qzv (306.7 KB)
The following deblur stats: deblur.qzv (480.1 KB).
I suggest that we need 2x230 bp Illumina runs for this primer system to use the denoising approach in the future.
I am glad, if you have other suggestions.
Thanks a lot!
above you said that the expected amplicon size is 394. Your sequences should be plenty long.
HOWEVER
I just discovered the problem here. Your sequences appear to have already been filtered and trimmed prior to attempting to merge/input to dada2. So the quality plots screnshot you showed above only represents a fraction of the reads; other reads are much shorter (they have been trimmed already! so dada2 can't do its job!). This will be an issue for dada2, vsearch, and deblur.
So essentially the problem is not with dada2 here, or with your read length, just that your read quality is evidently not quite good enough on this run (though I cannot be sure, as I do not know what sort of trimming was applied).
What type of trimming are you using before dada2, and why? Could you share the quality scores plot QZV of the untrimmed reads? (I do not think you shared this for the trimmed reads; could you please share that too?)
Your solutions are:
do not trim prior to dada2; we may be able to massage the data a little bit to get more length.
only use the forward reads for this analysis.
Those were a feature table summary, not the deblur stats (which are an output of the deblur denoise-16S method). I do not need that QZV now, since it looks like the problem is originating much earlier, but just for future reference.
{kind=link}
{kind=link}