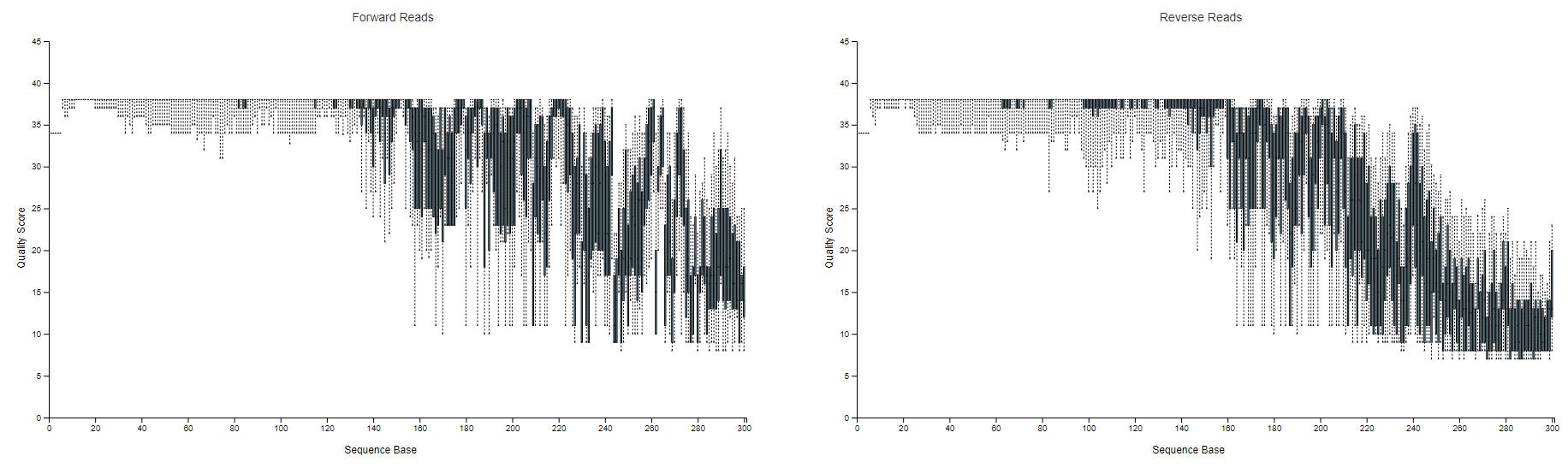
Hey @colinbrislawn. On this same topic, I wonder what you suggest when sequence quality drops off markedly before the R1 and R2 reads will overlap.
For example, check out this demux stats (online, and the raw file (312.8 KB)). These were 300 bp PE reads from a MiSeq, but quality really drops off after ~150 bp. If I truncate reads to 150bp, very few will overlap. If I truncate to 240bp so they overlap, very few will pass quality filters.
Is there ever a time where you would suggest analyzing these data as a SE run? E.g. only analyze the 150bp of the R1 reads?
Or, since my goal is simply to document presence of certain taxa, not to quantify any sort of abundance, I could even analyze the R1 reads and R2 separately, then just combine the final taxonomic identifications later.
The amplicon is 357 bp long, so with 300 bp PE sequencing, we should expect nearly total overlap without trimming. However, trimming down to 150-160bp with just the R1 would miss almost half the gene. If I do need to trim each read to 150bp, it seems like it would make sense to analyze both the R1 and the R2 separately (e.g. as two SE runs). Does that make sense to you?
Yes, that makes sense to me. With those quality scores, I think that's your only option.
(Maybe the sequencing core is willing to consider this a failed run and rerun it for you.)
...Unless you amplicons are much shorter than you expect. Is that possible?
I've seen read quality like than when we accidentally used a 250 bp paired-end kit to to sequence a <200 bp region of the 18S gene.
Thanks for your input! I'll give this a shot. Do you think it's worth combining the R1 and R2 reads (without merging them into one read) into one rep-seqs.qza file to train a taxonomy classifier/identify taxonomic units? Or just identify taxonomic units with the R1 and R2 reads separately?
I don't THINK that's what's happening here. We did sequence some other shorter amplicons on this run, so that may be happening there, but this cytb amplicon should be 357 bp. I'll look into it and talk with the sequencing core about the quality issues.
This is possible in theory, but in practice, there are many unexpected implications of working with an amplicon with a huge gap in the middle of it. Reviewer three will have many questions!
This is much easier, and probably more defensible.
Thanks for the updates! Let me know what you find,
Colin
Hey @colinbrislawn. After some digging, it seems that these quality issues are just due to the nature of the eDNA that the data were derived from. I was wondering if I might change the error acceptability threshold in the dada2 denoise step to possibly include more data. I see that --p-max-ee is set to 2 by default, but I'm not sure how this relates to the Phred scores that are kept or discarded. I assume I could increase --p-max-ee but I'm not sure what new values would be appropriate given the documentation. Any suggestions? Or should I just avoid this all together?
It might be worth trying a bunch of truncation settings to see if you can get these reads to join, or just use the forward read truncated around 150 or 200.