Hello Everyone,
So, I have successfully denoised my 16S V3–V4 paired-end reads from stool samples, my forward reads (FRs) are all high quality (median score 40) at upto 301bp length, but my reverse reads show poor quality (with lowest whiskers ~12–24 at the start and median between ~24–40). Here is a figure depicting the read qualities:
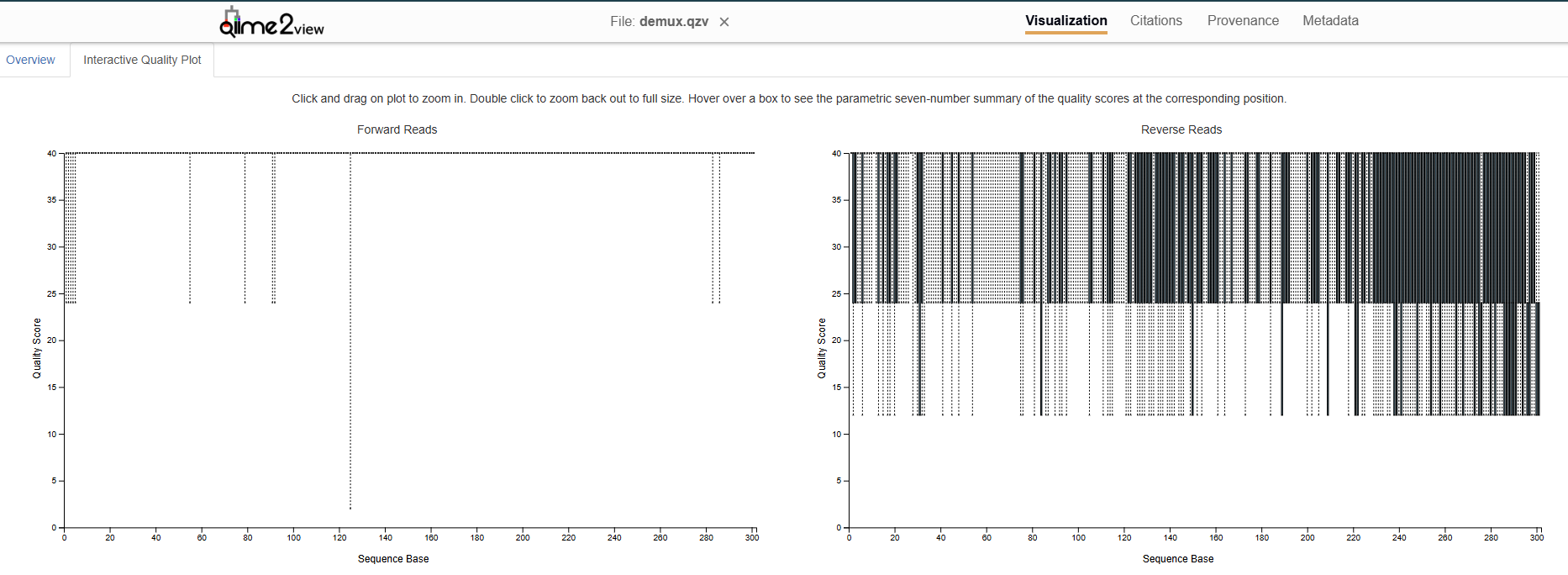
So, I’ve used following parameters for denoising using DADA2 and obtained the attached results:
qiime dada2 denoise-paired
--p-n-threads 8
--p-max-ee-f 1.50
--p-max-ee-r 1.50
--i-demultiplexed-seqs demux.qza
--p-trim-left-f 0
--p-trunc-len-f 301
--p-trim-left-r 0
--p-trunc-len-r 270
--o-representative-sequences asv-seqs.qza
--o-table asv-table.qza
--o-denoising-stats denoising-stats.qza
--verbose

I seem to be losing a lot of reads as chimera in DADA2. Therefore, I opted for deblur as well using the following parameters and obtained the attached results:
qiime vsearch merge-pairs
--i-demultiplexed-seqs demux.qza
--p-threads 4
--o-merged-sequences joined-paried-demux.qza
--o-unmerged-sequences leftover-unjoined.qza
qiime quality-filter q-score
--i-demux single-end-demux.qza
--o-filtered-sequences filtered-demux.qza
--o-filter-stats demux-filter-stats.qza
qiime demux summarize
--i-data filtered-paired-demux.qza
--o-visualization filtered-paired-demux.qzv
qiime deblur denoise-16S
--i-demultiplexed-seqs filtered-paired-demux.qza
--p-trim-length 301
--p-sample-stats
--o-representative-sequences rep-seqs-deblur.qza
--o-table table-deblur.qza
--o-stats deblur-stats.qza

I set my truncation length to 301bp (only the read length of the FR) in Deblur, but I was still losing a lot of data as chimera, having less than 10k reads-deblur per sample in most cases.
Since I have merged the paired-end sequencing reads. next I simply increased truncation length to 440 and got the attached results:
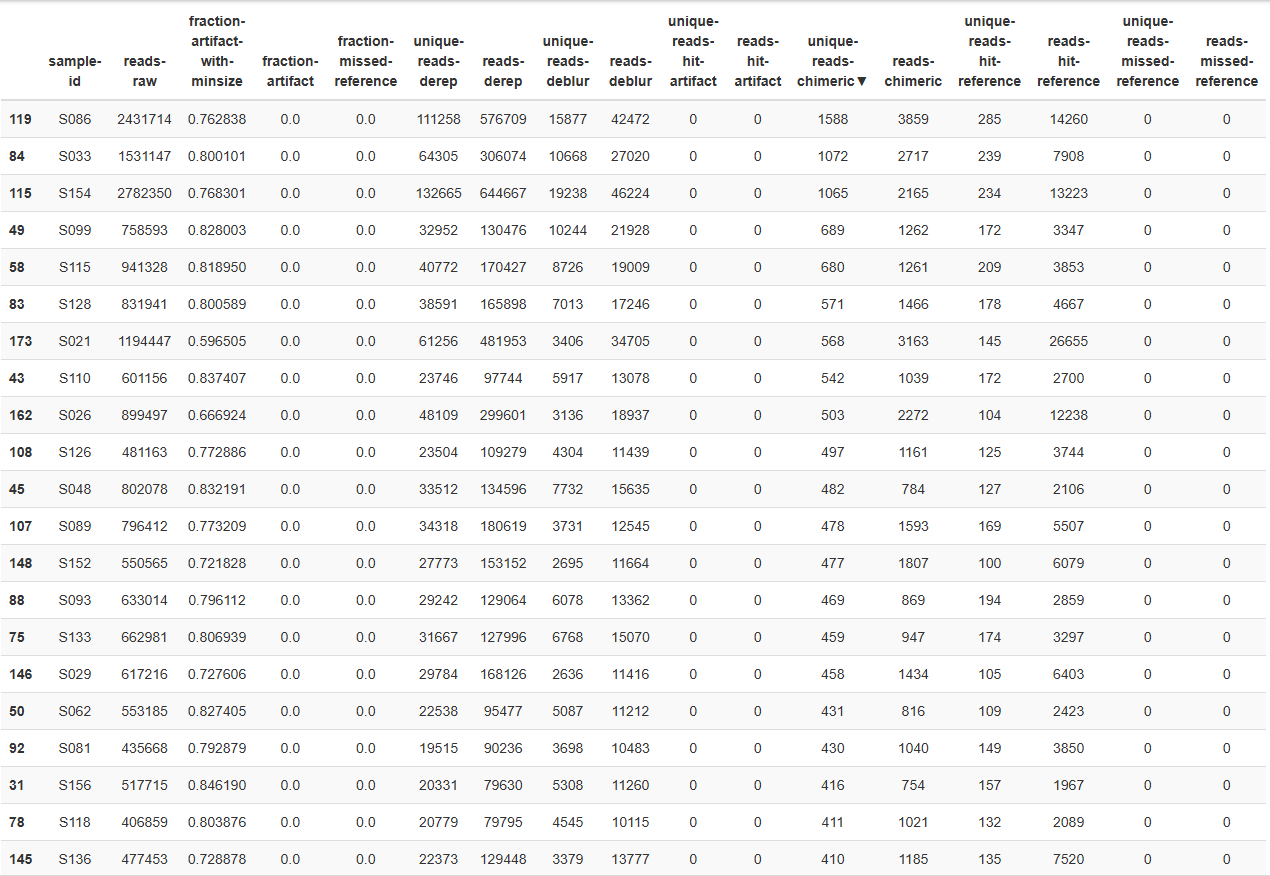
So, here I seem to be losing more reads than DADA2, mostly as artifacts. Therefore, having left with little to no reads for diversity analyze.
Now I am really at a loss and confused about how I can salvage the highest data for proceeding toward my next step of the analysis. Any suggestions or references for further reading will be greatly appreciated.