I ran into problems with taxonomy classification: I do get often a classification that has only 'unassigned' reads, but when I rerun the analysis often enough, I do get once in a while a classification at various taxonomic levels (as expected). To make it clear: I do not run different analyses, I just re-run the same command again and again until I get taxonomic classification. This is the command line:
In order to find out what is happening, I compared three sets of data:
A) a set of 28 samples
B) a different set of 14 samples
C) a combination of the sets A) and B)
Using the identical classifier file (silva_132_97_16S_341F-805R_classifier.qza) I get good taxonomc classification for case A) and B) with the command above, while I get only unassigned rep seqs in C).
Does anybody have an idea what might be wrong with the samples.
I attach the visualization file from 'qiime metadata tabulate' for inspection, and I can provide more details if necessary.
Thanks @arwqiime, it sounds like the feature classifier is being very badly behaved for you. Thanks for doing a bit of extra work to try to isolate the problem.
Are you please able to share the rep seqs and the classifier with us?
It would be great if I were able to reproduce the cases that change between runs, because that is something I would definitely like to fix.
Your other example with sets A, B, and C would be really useful, too.
Hi @BenKaehler,
here are the rep seqs and the classifier (as a download link).
Bact1-i_deblur is from the 28 sample data set (A)
Bact1-M_deblur is from the 14 sample data set (B)
Bact1-Mi_deblur is from the 42 sample data set (C)
I'm not sure what you mean with 'other example with A, B, and C'; please tell me which other data could be helpful to investigate.
In addition: I tried to run the analyses with differnt numbers of CPUs (1, 4, 10), same effect. I also tried to fugure out if '--p-reads-per-batch' would have any effect, but I could not find documentation on 'good number' or what range of reads to be precessed in a batch should be targeted.
Additional info: I processed amplikon reads from the same experiment with the identical workflow, but now with the fungal ITS as the amplified region and an ITS classifier, and it finished as expected!
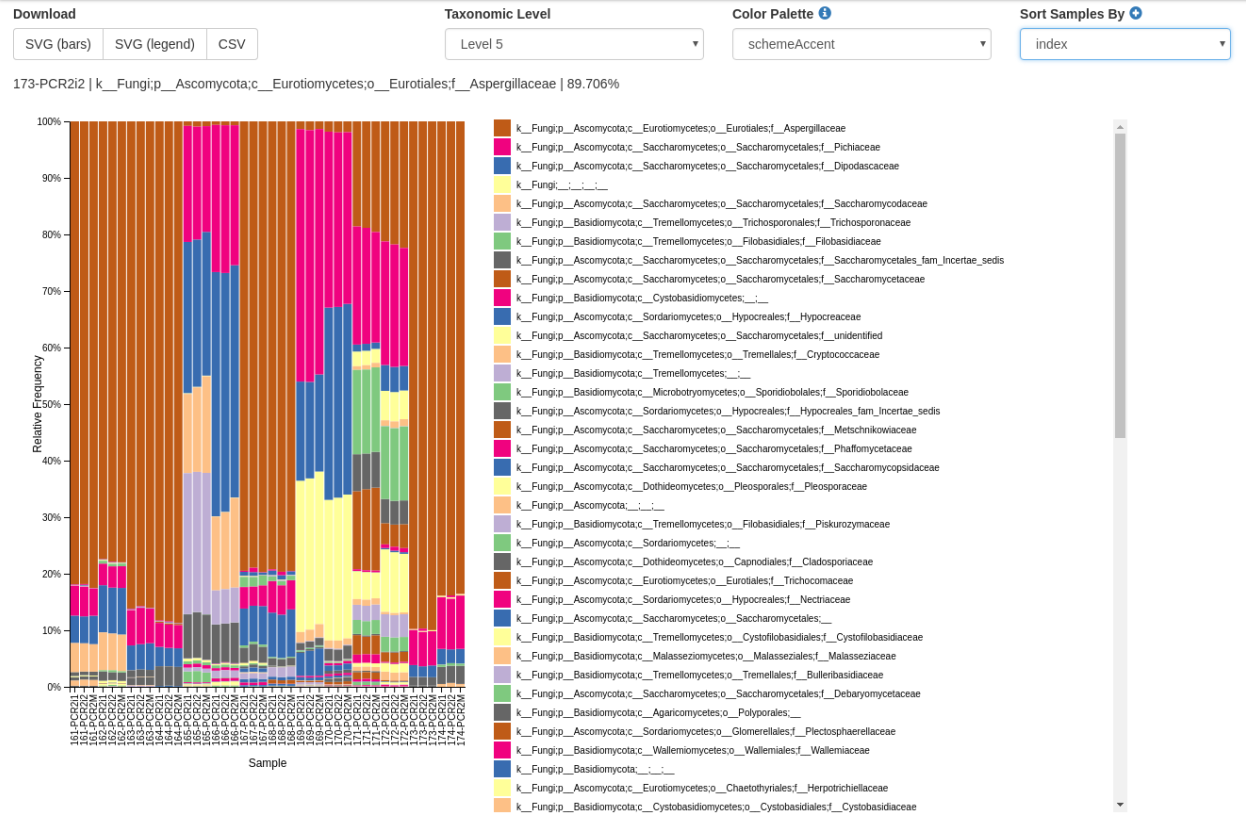
The screenshot showes the taxa barplot of the 42 sample set (corresponding to 'C' above, consisting of A (two independent runs on one machine) plus B (singe run on a different machine)).
So, I assume that my analysis workflow should be ok...
Hello!
I would like to add another short question related to the classify-sklearn output and the subsequent taxa bar-plot visualization:
We would like to screen our samples for pathogenic fungi and oomycetes. We generated amplicons with fungi specific primers as well as with modified primers known to amplify oomycota ITS regions in addition to fungal ITS regions. For taxonomic classification we used naive-bayes trained classifiers from UNITE 7.2. However, since these reference data sets contain only few oomycota sequences, we combined the UNITE data with oomycota data from BOLD (barcoding of life database) and generated a second 'combined UNITE-BOLD classifier'.
A classify-sklearn analysis followed by 'metadata tabulate' and 'taxa barplot' showed no oomycota reads in our dataset using the combined classifier. To test the oomycota classifier, I rerun the classify-sklearn analysis using a classifier consiting only of the BOLD oomycota data; here, I somehow 'forced' taxonomic annotations to oomycota sequences. Since this happend, I assumed that the classifiers were ok.
Since I could not identify oomycota reads in the csv table of the taxa barplot using the combined UNITE-BOLD classifier, I conclude that there are no oomycetes 'detectable' in my amplicon sample. I this conclusion correct?
If I want to test such non-standard classifiers, would it be advisable to add a certain number of known oomycota reads to the samples as 'spike-ins' to test the classifiers?
Hi, I ran into more problems with qiime feature-classifier classify-sklearn:
In addition to deblur, I wanted to analyse the ITS data (see taxa barplot from above) using
qiime vsearch cluster-features-open-reference ...
qiime vsearch uchime-denovo ...
qiime feature-table filter-features ...
Followed by
qiime feature-classifier classify-sklearn ...
However the last command resulted in memory errors (see: qiime2-q2cli-err-iy124g83.txt).
I updated from 2018.6. to 2018.8 and restarted classify-sklern and got a memory error, but shorter than the first one (see qiime2-q2cli-err-tkph9_5b.txt)
My machine hat the following installed;
KiB Mem : 49461372 total, 48903796 free, 371284 used, 186292 buff/cache
KiB Swap: 2097148 total, 1982504 free, 114644 used. 48729756 avail Mem
I assume, this should be enough!
Thanks @arwqiime, those are some really interesting experiments.
Are you using the "developer" UNITE sequences? The standard sequences come pre-trimmed so your special oomycota-specific regions may not be in the reference. See the tutorial for details.
Taxonomic classification of amplicons can only be as good as the information in the amplicons. Classification is not perfect. Our recent work has focussed on trying to resolve the inevitable ambiguities.
A classifier can only classify among the classes it has seen, which is why you were able to force classification by training on oomycota only.
You can't conclude that there are no detectable oomycetes in your sample, unfortunately. Spike-ins could help. A cheaper method would be to look at what the oomycetes were misclassified as (by comparing results from your BOLD and UNITE-BOLD classifiers) and eyeballing the reads, the reference sequences that they were misclassified as, and the oomycota reference sequences, to see if there are any obvious reasons for the confusion.
In relation to the memory issue, try running classify-sklearn with --p-n-jobs 1. If you've got lots of CPUs the classifier might be loading many large classifiers into memory.
Thanks @BenKaehler, I assume that I have asked too many question that led to some confusion. Let me try to break it up into independent questions:
No. 1: my initial question was related to a good taxonomic classification when using amplicon data from independent runs (indicated as A and B, respectively) and the surprise that when using all runs in one analysis (A+B=C) did not result in any taxonomic classification. The same classifier for bacteria (not fungi)(silva_132_97_16S_341F-805R_classifier.qza) was used.
The other question was related to ITS amplifications of the same experiment which gave me good taxonomic classification when using classification file made from UNITE developer sequences PLUS some BOLD sequences of ascomycota, but different from BOLD classifier alone. .
In fact, I wanted to use the full UNITE developer files: I imported the fasta file and used it together with the taxonomy artifact file to 'qiime feature-classifier fit-classifier-naive-bayes', but I received the following error message:
ValueError: Invalid character in sequence: b't'.
Valid characters: ['S', 'H', 'D', 'G', '.', 'N', 'W', 'Y', 'M', 'R', 'T', 'V', 'B', 'K', 'A', '-', 'C']
Note: Use lowercase if your sequence contains lowercase characters not in the sequence's alphabet.
I inspected the sequences in my default sequence analysis software (Geneious) and could not find errors. SInce the sequences were quite long, I decided to trim the sequences in Geneious using the primers used for amplification (ITS1F / ITS2; see screenshot below). Most sequences are too short in the SSU region and the ITS1F sequence could not be detected, but the ITS2 region could easily be detected and the downstream region be trimmed (red boxes). Therefore, I used the trimmed sequences, exported them as fasta, and constructed the classifiers described above.
From your reply today, I decied to test the original UNITE developer sequences, too. In order to avoid the error above, I imported the fasta into Geneious and re-exported them again as fasta without any modification. When using this re-exported fasta file in q2 and make a classifier, the error message is gone. No idea what might have gone wrong, but the 'original' UNITE fasta file does not work in my hands...
Hi @arwqiime,
We are still looking into your initial question — sorry for the delay on this but we have been backed up this week and this is a complex problem. We will keep you posted.
I can answer your other questions though:
It sounds like BOLD must contain sequences covering clades not covered by UNITE, and vice versa. So they complement each other and improve performance on your dataset.
We've run into this before — the developer sequences do contain some lowercase characters. This causes problems with scikit-bio, the program QIIME 2 uses for handling these sequence data.
The fix is easy — just find and replace lowercase with uppercase using your preferred method.
Based on the fact that the problem disappeared after exporting from Geneious, my guess is this program already converts lower->uppercase on import.
Note that you can also do this trimming within QIIME 2 with feature-classifier extract-reads — just noting in case you are curious (the only advantage is then the trimming would be stored in provenance so you would have a better record of the workflow you used)
It really is just lowercase characters in the sequences. Very silly problem to have but an easy fix (and we have an open issue somewhere to streamline this by converting lowercase->uppercase on import)
I hope that helps! Stay tuned for more details on problem #1!
@arwqiime,
I think I have a pretty good guess what's going on, but I will need you to run some tests to confirm.
classify-sklearn should not produce stochastic results like you describe — unless if the classifier is getting confused about the orientation that your reads are in. Read orientation can be set manually using the --p-read-orientation parameter. If this is not manually set, the classifier will attempt to determine the read orientation based on the first 100 reads or so (can't remember the exact #).
This almost always works — unless if the reads are in mixed orientations or if the reads contain non-biological sequence information (or at least sequence information not contained in the reference sequences, e.g., primers that were not trimmed from the query).
Could you try the following?
Re-run classify-sklearn with --p-read-orientation set to same and set to reverse-complement. Please post the QZVs of the results here and let us know if that is a clear fix! No need to proceed to steps 2/3 if setting the orientation manually works.
Confirm that your sequences are not in mixed orientations (it looks like they are not, based on cursory examination, but I do not know your sequencing protocol so pls let us know if they are mixed).
Confirm that your query sequences are trimmed to the same sites as your reference sequences and do not contain untrimmed primers or non-biological DNA that is not present in the reference sequences.
If that doesn't work, there could be an issue with the classifier that you trained, but that seems less likely at this moment.
Please give that a try and let us know if that solves this issue!
Hi @Nicholas_Bokulich,
Great! The '--p-read-orientation same` seemed to solve my problem. See attached visualization.
Nevertheless, I will inspect my read for any non-biological sequences; they should be free of primer sequences, but I will see whether this is really true.