Hello,
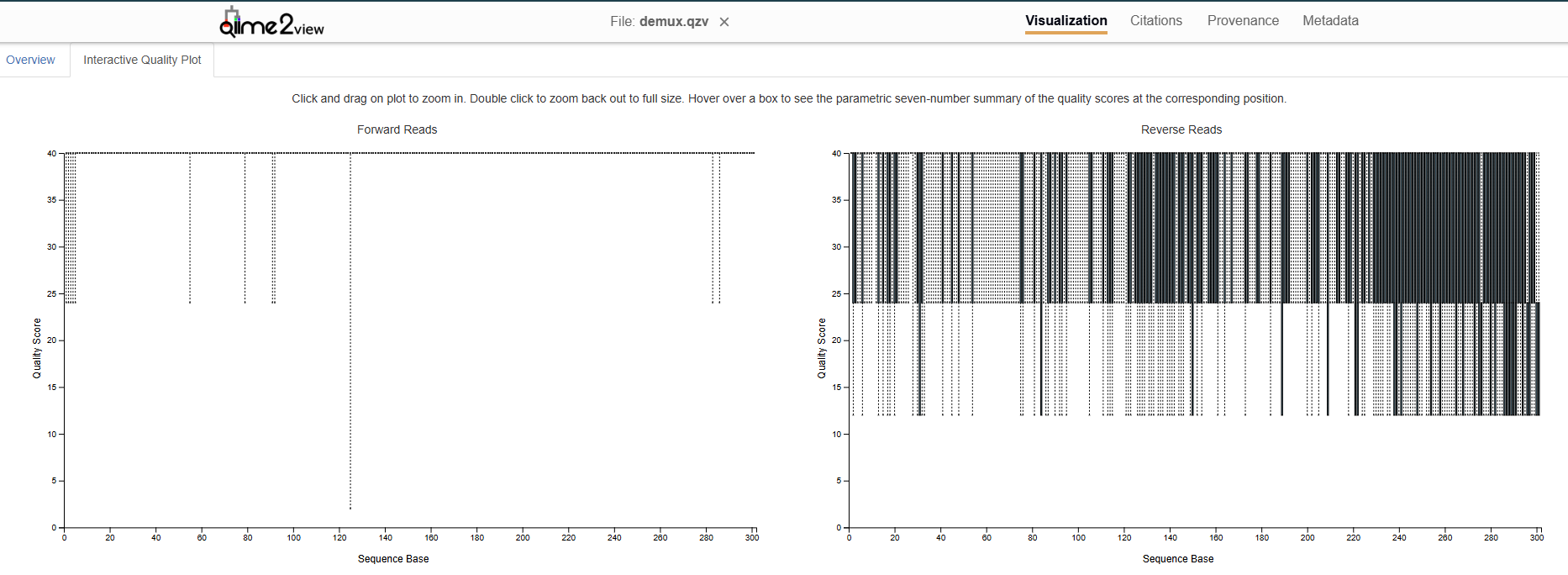
I’m working with 16S V3–V4 paired-end reads, and my reverse reads show poor quality (with lowest whiskers ~12–24 at the start and median between ~24–40), so I’m deciding between using denoise-single (forward reads only) or truncating the reverse to ~230–270 where quality noticeably degrades. Could you advise me on best practice for this scenario?
It’s been running for 4 days. The demux.qza is ~28 GB, and Task Manager shows 4 CPU cores allocated to vmmemwsl. I’ve attached the sequence count summary.
Is this runtime normal for a dataset of this size?
Would you recommend stopping and re-running with --verbose to monitor progress?
If paired-end denoising is feasible, what truncation settings would you suggest to maintain sufficient overlap for V3–V4 while minimizing low-quality bases?
I don't think that runtime is particularly unreasonable for this. These things can take a long time. If you want things to run faster, and you have the RAM for it, you can pass an argument to the --p-n-threads parameter in the future.
As far as what to do about your reverse reads, if you can't get them re-sequenced, then unfortunately, going with only the forward may be the most sensible option here even though you will most likely be losing one of your variable regions by doing so.
Do any other @moderators have better advice on this?
My recommendation would also be to drop the low quality reverse reads and move on with the forward reads only. It’s going to be hard to rescue that data, and you’ll likely land on a position where you’re sacrificing otherwise high quality reads (samples!) for sequences you’re less confident in.
I tend to think of the balance in three ways:
More samples give me more power to characterize the thing I’m interested in. (More statistical power! More fun!!!)
This is always my goal.
More reads, more potential information for each sample. I’m less likely to drop a sample I might have otherwise saved, but I’m also more likely to detect prevelant but low abundance taxa. So, I get more insight into the mechanism
I balance this with #1 and sometimes #3
More read length, more taxonomic specificity. A read that’s longer will contain more information to distinguish bacteria. If there’s a difference between two species in your amplicon region at nt 200 from the primer and your read is 100nt long, you wont ever be able to separate those taxa.
At the same time, my ability to distinguish taxa means nothing if the data supporting that assignment is poor or I dont have enough samples to do my statistics.
The other thing I noticed is thsi:
Based on the size of your demux, I also want to check that this isn’t a metagenomic data? I tend to have much smaller demux files with my 16S.
If it’s multiple runs combined, you should process those seperately in DADA2 (use the same parameters across all runs). If it’s metagenomic data, consider a different annotation pipeline.
If it is 16S, check your sample number. The rate at which DADA2 learns error is associated with the number of samples and IIRC, lower sample numbers lead to poor performance.
Thank you both @Oddant1 and @jwdebelius for your insights and valuable suggestions.
For your questions, yes, this is metagenomic data from 178 samples (V3-V4 region of 16S amplicon, as mentioned earlier). This is data from a single run, and I'll be adding data from more runs in the future.
Thanks for the advice. May I know what different annotation pipeline you’re implying? Is it deblur? because I read in the forum that for multiple runs combined, using Deblur (which uses static error model) is more appropriate.
Also, since Deblur uses only forward-read, as far as I know, are there any additional steps that I have to perform when using Deblur instead of DADA2?
Apparently I can’t read until I have more tea ! I just wanted to check, because that’s a lot of data for a 16S run! You’re in the right pipeline overall.
In terms of denosing algorithms, I think both DADA2 and Deblur have advantages. DADA2 tends to outperform Deblur most microbiome related performance metrics in the independent comparisons. (See: Nearing et al, 2018, Proden et al, 2020; Fares et al, 2025; although it's worth noting that not all papers ran the default pipelines and some results maybe influenced by ignoring key algorithmic assumptions.)
The benchmarks agree that DADA2 suffers in terms of computational performance. It is by far the slowest of the denoising algorithms because it learns an error rate per sequencing run, while Deblur uses an assumed upper limit error rate. (Unoise3, which is the other method mentioned in the articles but not available in QIIME 2 has a partitioning algorithm and tends to be the fastest.) So, is that enough reason to switch to Deblur? I dont know. I find I get the best performance with Deblur when I use single end 150nt reads amplified with the 515F-806R primers; this has to do with how the algorithm was trained. I have used it on other primer pairs, but I lose a lot of reads with that processing.
The other option to consider might be to look into additional computational resources. This may not be soemthing you can process on your laptop, and you may need to look into additional compute from a local high performance computer (some insitutitions or jursitictions have them) or commercial source.