Thanks for explaining, and thank you for your patience, @YuZhang. I think you have a solid understanding of what's going on here. Depending on your study, you may be suffering from a desire to optimize unnecessarily.
Results so far
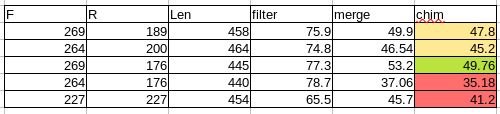
Of the examples you show above, your best parameters look like the ones shown in stats-dada2-2.qzv, where you capture almost 50% of your sequences. Why? You retain the most data, and probably don't bias your data in the merge process.
Always pad your estimated amplicon length
One important insight from this table: when your sequence length drops to 440, you lose many more sequences in merging.
Your math here looks correct to me (sorry for any confusion I may have caused above - I overlooked that you had pre-trimmed primers). However,
many of your sequences are probably slightly longer than 428 bp. If you truncate to a total length of 428 + 12 = 440, these sequences will fail to merge.
Failing to account for sequence length variation can bias your data by dropping any sequences that are naturally a little longer, potentially disproportionately impacting certain taxa. A literature search may be necessary to figure out how much variation in length to expect in between 338 and 806. If in doubt, it's probably better to lose sequences to filtering than to merge failure.
Is this good enough?
With your best parameter set above, you have over 18,000 sequences in the shallowest of your samples. For many studies, that's more than enough depth for successful analysis.
Sequence attrition from denoising can be stressful, but it's important to remember what we're doing during this process. Quality filtering drops untrustworthy reads. Denoising with DADA2 corrects badly-read positions, and chimera-checking removes sequencing chimeras. This conservative approach helps remove artifactual taxa from your data, and generally leaves you with a better representation of the biological community you're studying at the "cost" of a bunch of untrustworthy reads.
Can you do better?
Maybe! If your study requires greater sequencing depth, you can continue to tweak these parameters until you get a better retention rate, at the cost of work and compute time. Focus on truncating the lowest-quality positions while preserving an adequate overall length and you'll be fine.
You could also denoise only your forward reads, and trade read length for some additional sequencing depth by skipping the merge process. If you're losing 25% in merging, that could be a significant improvement, but only if you don't need your full target amplicon.
Though I don't recommend it in the general case, it is possible to make DADA2 run more permissively if your data requires it. The q2-dada2 and DADA2 docs will be your best guides, along with forum posts here. Again, DADA2 has sensible defaults for most 16s work, so consider whether your study needs this.
Good luck, and let us know how everything goes!
Chris