I extract 16S seqs from metagenome seq data. Then I use qiime(qiime2-2019.7) to analysis. I encountered error when running dada2 denoise-paired step.
here is my command:
qiime dada2 denoise-paired
--i-demultiplexed-seqs ./meta3_16s.qza \
--p-trunc-len-f 140 \
--p-trunc-len-r 138 \
--o-representative-sequences ./rep-seqs-dada2.qza \
--o-table ./table-dada2.qza \
--o-denoising-stats ./denoising-stats.qza \
--output-dir ./temp \
--p-n-threads 6 \
--verbose
This returns the following error:
Running external command line application(s). This may print messages to stdout and/or stderr.
The command(s) being run are below. These commands cannot be manually re-run as they will depend on temporary files that no longer exist.Command: run_dada_paired.R /tmp/tmp_v9xvd13/forward /tmp/tmp_v9xvd13/reverse /tmp/tmp_v9xvd13/output.tsv.biom /tmp/tmp_v9xvd13/track.tsv /tmp/tmp_v9xvd13/filt_f /tmp/tmp_v9xvd13/filt_r 140 138 0 0 2.0 2.0 2 consensus 1.0 6 1000000
R version 3.5.1 (2018-07-02)
Loading required package: Rcpp
DADA2: 1.10.0 / Rcpp: 1.0.2 / RcppParallel: 4.4.3
- Filtering ..........................................
- Learning Error Rates
470646820 total bases in 3361763 reads from 1 samples will be used for learning the error rates.
Error in err[c(1, 6, 11, 16), ] <- 1 :
incorrect number of subscripts on matrix
Execution halted
Traceback (most recent call last):
File "/home/luohx/software/anaconda3/envs/qiime2-2019.7/lib/python3.6/site-packages/q2_dada2/_denoise.py", line 234, in denoise_paired
run_commands([cmd])
File "/home/luohx/software/anaconda3/envs/qiime2-2019.7/lib/python3.6/site-packages/q2_dada2/_denoise.py", line 36, in run_commands
subprocess.run(cmd, check=True)
File "/home/luohx/software/anaconda3/envs/qiime2-2019.7/lib/python3.6/subprocess.py", line 418, in run
output=stdout, stderr=stderr)
subprocess.CalledProcessError: Command '['run_dada_paired.R', '/tmp/tmp_v9xvd13/forward', '/tmp/tmp_v9xvd13/reverse', '/tmp/tmp_v9xvd13/output.tsv.biom', '/tmp/tmp_v9xvd13/track.tsv', '/tmp/tmp_v9xvd13/filt_f', '/tmp/tmp_v9xvd13/filt_r', '140', '138', '0', '0', '2.0', '2.0', '2', 'consensus', '1.0', '6', '1000000']' returned non-zero exit status 1.During handling of the above exception, another exception occurred:
Traceback (most recent call last):
File "/home/luohx/software/anaconda3/envs/qiime2-2019.7/lib/python3.6/site-packages/q2cli/commands.py", line 327, in call
results = action(**arguments)
File "</home/luohx/software/anaconda3/envs/qiime2-2019.7/lib/python3.6/site-packages/decorator.py:decorator-gen-459>", line 2, in denoise_paired
File "/home/luohx/software/anaconda3/envs/qiime2-2019.7/lib/python3.6/site-packages/qiime2/sdk/action.py", line 240, in bound_callable
output_types, provenance)
File "/home/luohx/software/anaconda3/envs/qiime2-2019.7/lib/python3.6/site-packages/qiime2/sdk/action.py", line 383, in callable_executor
output_views = self._callable(**view_args)
File "/home/luohx/software/anaconda3/envs/qiime2-2019.7/lib/python3.6/site-packages/q2_dada2/_denoise.py", line 249, in denoise_paired
" and stderr to learn more." % e.returncode)
Exception: An error was encountered while running DADA2 in R (return code 1), please inspect stdout and stderr to learn more.Plugin error from dada2:
An error was encountered while running DADA2 in R (return code 1), please inspect stdout and stderr to learn more.
See above for debug info.
besides this error, I also have other doubts.
-
- when not use the parameter --verbose, the error return like as follow, but I counld not find the file /tmp/qiime2-q2cli-err-04x_31km.log:
Plugin error from dada2:
An error was encountered while running DADA2 in R (return code 1), please inspect stdout and stderr to learn more.
Debug info has been saved to /tmp/qiime2-q2cli-err-04x_31km.log
-
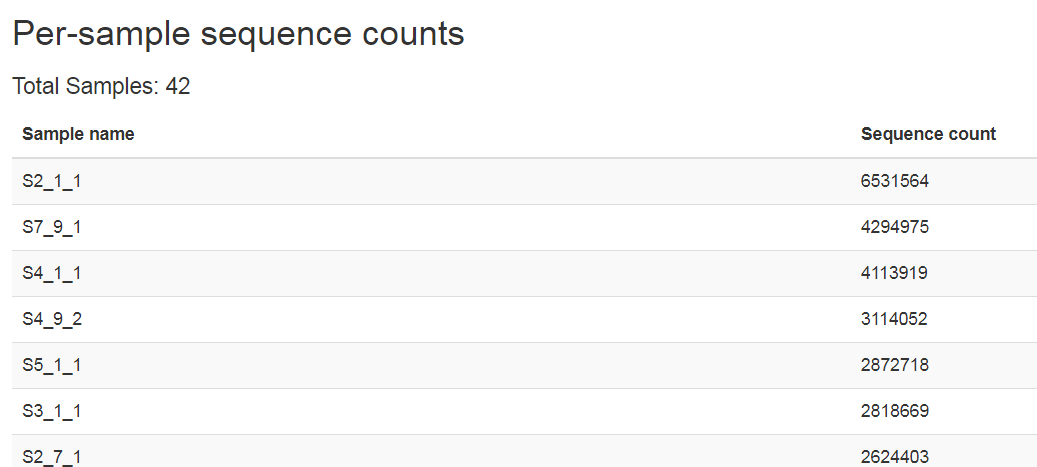
- I have imported 42 pairend fastq.gz 16S seq data, why in the error message there are only one sample?
- I have imported 42 pairend fastq.gz 16S seq data, why in the error message there are only one sample?
-
- the following is the quality plot of the importing 16S data. Because It is extracted from metagenome, the length is 150nt. I am not sure the overlap length between the forward and reverse reads, and want to maintain as long as possible reads, so the truncating parameters are loose, set
--p-trunc-len-f 140 \--p-trunc-len-r 138 \. And I trimmed the primers and barcodes bytrim galore, I don't use the parameter--p-trim. I doubt whether the low seq quality is the reason of the error.
- the following is the quality plot of the importing 16S data. Because It is extracted from metagenome, the length is 150nt. I am not sure the overlap length between the forward and reverse reads, and want to maintain as long as possible reads, so the truncating parameters are loose, set


I have search the problem in qiime2 forum, there are some similar topics. But I go through the error detail, there are not exactly the same. so anyone can figure out what's the problem and how can I correct it . Many thanks