Dear community,
We are dealing with 16S Pacbio CCS sequences (produced using Pacbio Revio system) but, at the denoising step, we cannot fully understand how dada2 filters the reads.
We tried different strategies to understand what was happening during the pipeline. We herein provide the commands used and the obtained resulting statistics using just one library as an example:
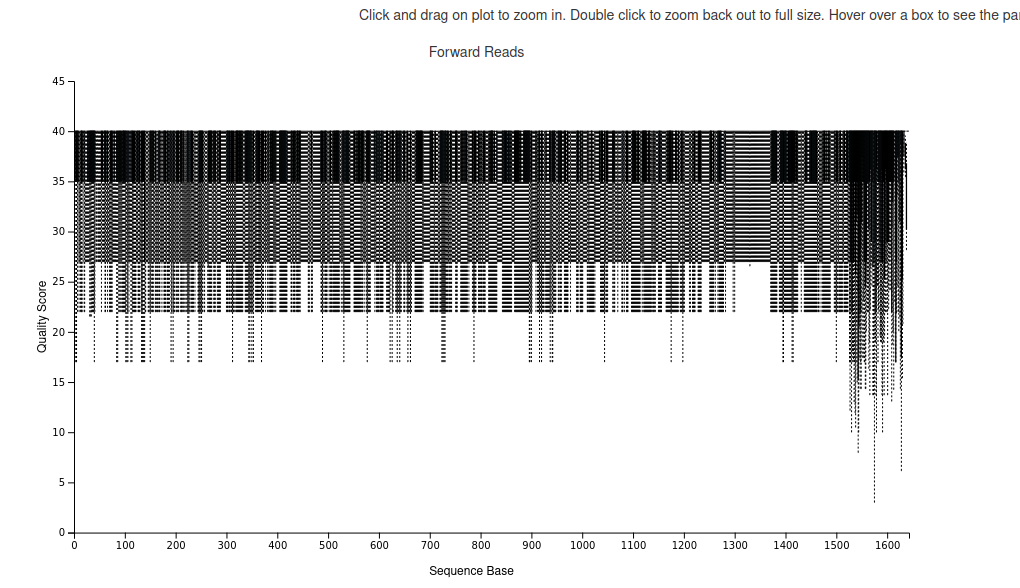
fastqc L3CTD.fq.gz

as you can see, the overall quality of the library is good. We hence decided to directly import the sequences in qiime2:
qiime tools import \
--type 'SampleData[SequencesWithQuality]' \
--input-path sample_metadata \
--output-path L3CTD.qza \
--input-format SingleEndFastqManifestPhred33
and process them using the dada2-plugin designed for CCS sequences without trim for quality (just for length):
qiime dada2 denoise-ccs \
--i-demultiplexed-seqs L3CTD.qza \
--p-front AGRGTTTGATYNTGGCTCAG \
--p-adapter TASGGHTACCTTGTTASGACTT \
--p-min-len 1200 \
--output-dir Denoise_dada2
qiime metadata tabulate \
--m-input-file Denoise_dada2/denoising_stats.qza \
--o-visualization Denoise_dada2/denoising_stats.qzv
As you can see primers are removed efficiently but, at the filtering step, many sequences are lost (only the 11% were retained).

At this point we would like to know why we were loosing a huge number or reads during this step (we didn't say to dada2 to remove reads depending on quality). As such we played with the expected error parameter of the plugin, raising it up to 10:
qiime dada2 denoise-ccs \
--i-demultiplexed-seqs L3CTD.qza \
--p-front AGRGTTTGATYNTGGCTCAG \
--p-adapter TASGGHTACCTTGTTASGACTT \
--p-min-len 1200 \
--p-max-ee 10 \
--output-dir Denoise_dada2
but the ouput was grossly the same .....

At this point, following this issue, we tried to filter the reads directly in R using the dada2 library. Here the commands:
# libraries
library(dada2)
library(Biostrings)
library(ShortRead)
library(ggplot2)
library(reshape2)
library(gridExtra)
library(phyloseq)
# path to files
file_raw <- "~/Scrivania/Felice/L3CTD.fq.gz"
file_noprim <- "~/Scrivania/Felice/L3CTD.fq.no_primers.gz"
file_filt <- "~/Scrivania/Felice/L3CTD.fq.no_primers.filtered.gz"
# oligonucleotides
primer_fw <- "AGRGTTTGATYNTGGCTCAG"
primer_rv <- "TASGGHTACCTTGTTASGACTT"
# reverse and complement function
rc <- dada2:::rc
# remove the primers
prim2 <- removePrimers(file_raw, file_noprim, primer.fwd=primer_fw, primer.rev=dada2:::rc(primer_rv), orient=TRUE)c
print(prim2)
# reads.in reads.out
#L3CTD.fq.gz 20613 18682
# check the lengths
length <- nchar(getSequences(file_noprim))
hist(length, 100)
# Filter with similar parameters used in the first denoising test in qiime2 dada2-denoising-ccs (see above)
track2 <- filterAndTrim(file_noprim, file_filt, minQ=0, minLen=1200, maxN=0, rm.phix=FALSE, maxEE=2, truncQ=0)
print(track2)
# reads.in reads.out
#L3CTD.fq.no_prim.gz 18682 5040
plotQualityProfile(file_filt)

As you can see, by importing the library in R and process it with dada2 functions we obtained:
- an equal number of primer trimmed reads with respect to previous qiime2 output
- a different number of filtered reads with respect to previous qiime2 output
5040 vs. 2345 (even using the same parameters configuration as we used at the first step in qiime dada2-denoising-ccs)
By adjusting the expect error value, as our second try, we got a very high discrepancy if comparing qiime2 output with R output (2357 vs 12097):
....
track3 <- filterAndTrim(file_noprim, file_filt, minQ=0, minLen=1200, maxN=0, rm.phix=FALSE, maxEE=10, truncQ=0)
print(track3)
# reads.in reads.out
#L3CTD.fq.no_prim.gz 18682 12097
plotQualityProfile(file_filt)

Our final remarks about this possible issue we bumped in are:
-
There is a high discrepancy in the filtered output when using dada2 within the qiime environment and the R release. How is that possible? Why dada2 in qiime is more restrictive in filtering with respect to dada2
filterAndTrimfunction? Isn't q2-dada2 implemented depending on R-dada2? Since qiime2 was designed to facilitate metagenomic pipelines, wouldn't it be better to improve this plugin? -
How we can afford this problem? It's not a big deal move all the libraries in R and then import them back to qiime, but if you have a better (and faster?) advice ....
-
Due to the overall good qualities of this library, shall we proceed with a minimum filtering using the maxEE=10 option or do you think we must use a more conservative approach (lowering the maxEE value)?
Thanks for the help!
Bests,
Claudio