I am new to qiime2 and microbiome analysis in general. I was interested in my unweighted unifrac result from qiime2 because significance testing with PERMANOVA showed pairwise differences all throughout my 3 groups. So I wanted to reproduce the results in R: I used phyloseq to do a PCoA plot (which was more or less the same as the q2 output, just a little different %ages in axes), but when I performed significance testing with pairwise adonis (as I learned was the equivalent of PERMANOVA in R) on the distance matrix, the p values I got were different such that 1 pairwise comparison was no longer significant.
So I tried running pairwise adonis again in PERMANOVA /but/ using the exported distance matrix from unweighted unifrac (with formatting modifications) result in qiime2, and I got significant results for all 3 pairwise comparisons.
My question is, why is the distance matrix from qiime2 different from that produced in phyloseq in R (phyloseq::distance)?
And,
would it be all right to use the phyloseq output for PCoA (using phyloseq ordinate function on physeq object) together with the adonis pairwise output run on the exported distance matrix of the qiime2 output?
Thanks for posting on the forums. Welcome to Qiime 2! :qiime2:
In order to track down the source of this issue, we need to compare the output files at each stage in this process. This could be due to different default settings, or very small differences that have a large effect. For example, in that form post you listed, one source of the difference was how Qiime 1 and R parse sample names! It could be any number of things.
That's a great place to start. What is the exact steps you used to make the distance matrix in R and Qiime? How do you know they are different?
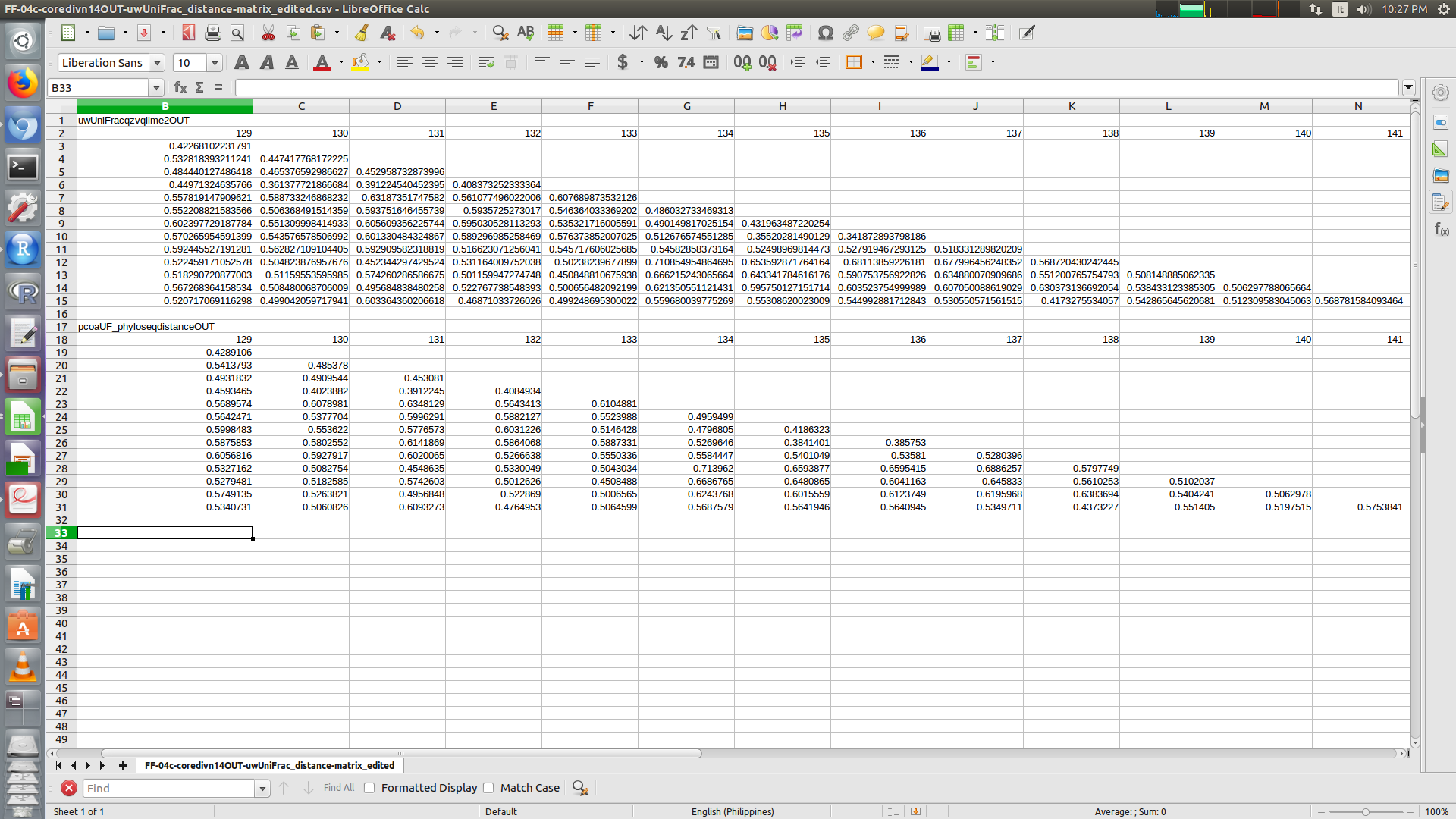
Okay, here are the two distance matrices. The first one is the qiime 2 output for unweighted UniFrac from the qiime diversity core-metrics-phylogenetic command; the second is the output from running phyloseq::distance in R on a physeq object (created by reading the exported tree, taxonomy ,metadata,feature table QZAs using the Qiime2R package).
Even if you generate these distance matrices in the same way in QIIME 2 vs. phyloseq (same metric, rarefaction, same rarefaction depth), there will be differences. The reason is that rarefaction (used by default in QIIME 2's core-metrics-phylogenetic pipeline, and not used by default in phyloseq) randomly subsamples your feature table — do that 10 times and you will retrieve 10 slightly different results since the subsample is random.
The fact that you see significant effects with QIIME 2 but not phyloseq is either due to this random subsampling, or more likely because in phyloseq you are not subsampling or normalizing at all (I am assuming since you have not mentioned normalization in phyloseq), so fairly large differences between these distance matrices may be expected. Unweighted unifrac will be very sensitive to sampling depth differences, which should be normalized across samples prior to unifrac (e.g., by rarefaction or another normalization method).
But for this purpose I would just like to use R for visualizing qiime2 data. For future reference I would just like to post how I found a way to use the distance matrix from qiime2 with the phyloseq ordinate command by converting the exported qiime 2 unifrac output into a dist-object and then using it for the distance argument in the phyloseq ordinate function. I have yet to see if it works, though.