Hello everyone!
I am new to this kind of tools so not sure what is the issue I have.
I would be grateful for any help.
I have two files from ILLUMINA Miseq and metadata file with barcodes, primers and etc. (the last file I did in table than changed the format to .tsv)
I tried a few ways to import the data but it usually something wrong with my commands or files. Please help me to figure out what I do incorrectly?
Thank you.
Is this post about a User Support Question? Those include questions about specific results while running QIIME 2, warnings observed while running a QIIME 2 command. Please do not post questions here that have to do with interpretation of results, general discussion, or technical support.
Before posting, please make sure you have the following information available, in order for us to help you in a timely manner:
Have you searched for the problem on the forum? It is rare that we see a new question asked, so make sure you do your homework before asking for us to commit our time to helping you.
Thanks so much for posting those screenshots, they were very helpful! It looks like you are missing one of the required files for your selected datatype, based on this error message that you are receiving:
Missing one or more files for EMPPairedEndDirFmt: 'forward.fastq.gz'
From the Importing Data tutorial, you'll see that for EMPPairedEndSequences you'll need .fastq.gz files for your forward and reverse reads, as well as for your barcodes.
Make sure all three of those files are saved within your emp-paired-end-sequences directory, according to the instructions in that tutorial. That should get you up and running here, but please let me know if you are still running into any issues!
Hello Liz!
Thank you for your response.
I created the directory named it emp-paired-end-sequences, then using the "mv" command I tried to move three files (metadata.tsv, forward.fastq.gz and forward.fastq.gz) they moved but zipped in one file with the same name emp-paired-end-sequences.
I tried to import using the commands, but it was the same mistake, then I tried to import it from the directory again not working.
I am sorry I don't know what I do wrong.
Thanks for giving that a try! If you take a closer look at the instructions in the tutorial I provided in my previous response, you'll see that you need to label your files as follows:
Barcodes: barcodes.fastq.gz
Forward Reads: forward.fastq.gz
Reverse Reads: reverse.fastq.gz
From the details and screenshot you provided in your most recent response, it looks like your emp-paired-end-sequences directory contains two subdirectories, named emp-paired-end-sequences and emp-paired-end-sequences (1).
I'd recommend just starting from scratch to prevent any further confusion - go ahead and delete these directories (you can do so with the rm -rf command before your directory name) and then start over with a new directory with the same name that contains all three of the files I discussed above.
Hopefully this helps clear up some confusion on this - let me know if you run into any further issues!
Hello Liz.
I am sorry if I confused you with the picture.
These two files is not subdirectories. Its files that ubuntu zipped instead of moving them, you can see two files, one zip, another unzipped. It should be three files as I mentioned above.
Thanks for providing that clarification! The three files that are required for this type of import are forward, reverse, and barcode reads. If you do not have a barcode read file, your import will not be successful. If possible, I'd recommend reaching out to your sequence provider and requesting this file for your forward and reverse reads.
Hello
Thank you
I can't do that, because we have the Illumina MiSeq in our institution and we did the sequencing by ourselves.
So, I can't do anything with our data?
Your data is still usable! You just won't be able to import it as an EMPPairedEndSequences datatype. Here are a couple of suggestions:
Depending on what your analysis needs are, cutadapt may meet your needs as it deals with adapters in sequence data.
Another option would be to look into bcl2fastq (previously known as CASAVA), which is the Illumina program used for demultiplexing - which may be appropriate for your dataset.
Hello Liz!
Thank you/
I thought that I could use Multiplexed paired-end FASTQ with barcodes in sequence¶ protocol
Our data format seems to be valid for this particular protocol
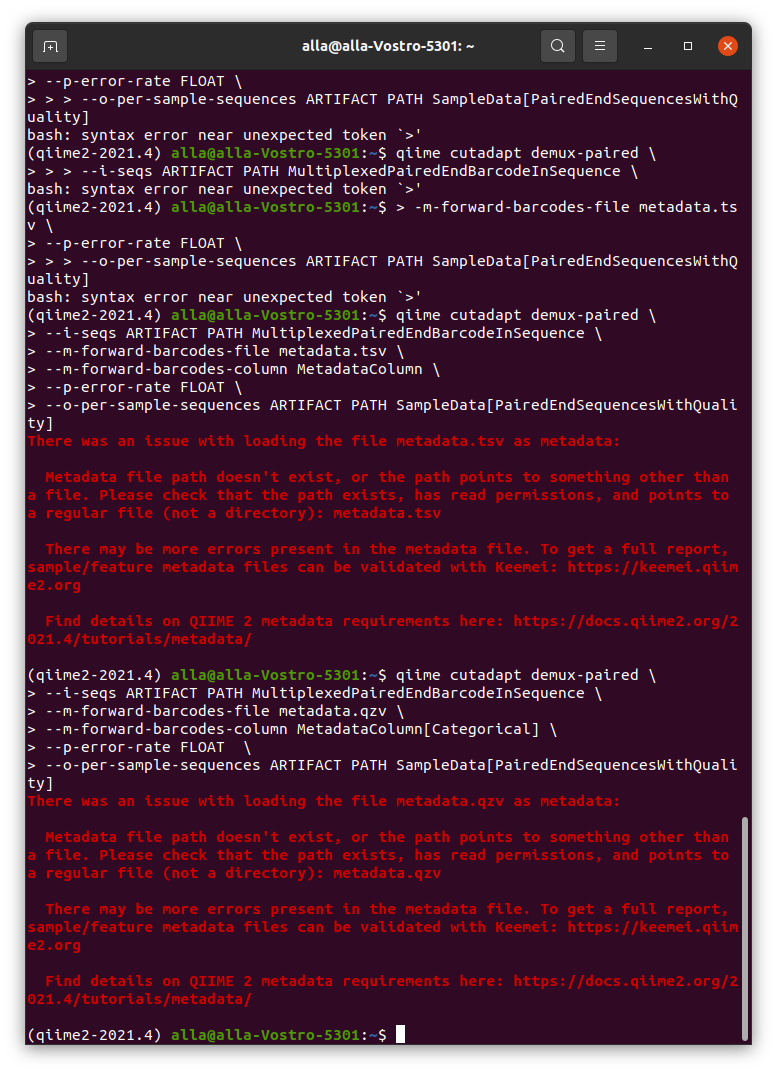
I actually was capable to import the fastq file when i deleted metadata file, this is pretty strange because the keemi keep saying that my metadata table is fine.
But I think i can't do anything without metadata file
Great catch! That should work for your use-case. However, from the first screenshot that you shared, it looks like you are attempting to utilize MultiplexedSingleEndBarcodeInSequence as your datatype, rather than MultiplexedPairedEndBarcodeInSequence.
I recommend going back through and running qiime tools import with MultiplexedPairedEndBarcodeInSequence as your format, and then making sure you are including your forward and reverse reads in your input commands.
In the second screenshot you provided, it looks like your metadata file wasn't found. When using qiime cutadapt demux-paired, you need to include your metadata file in your input commands. I know you mentioned that you deleted your metadata file, in which case I'd make sure you have that restored so that you can utilize it when running this analysis.
One final note I'd like to add is that you'll want to make sure you have your forward and reverse read barcodes in separate columns for q2-cutadapt, since you are working with paired end reads.





{kind=link}