Unfortunately, there was an issue and we ended up sequencing PE150. For analysis using QIIME2, I took two approaches: 1) treat as single-end, and 2) for merge using DADA2 justConcatenate. After denoising, I loose a lot of sequences (on average, only 10% of entire sequence is recovered).
My questions are:
Is it okay to assume that such low recovery rate after denoising is due to short sequencing reads? (after trimming, a get 110bp reads)
My sequencing depth was 1M per sample, so even with only 10% recovery, I deemed I have enough reads to proceed with the analysis (my rarefraction graphs also seemed fine). Would it be okay to infer information from these data?
If I understand correctly, DADA2 just Concatenate only works through the R package (not within QIIME2). After merging and preprocessing, I get two files: ASV table as a .txt file and feature sequence file as .fna. What is the best way to import the ASV table to QIIME2? I I tried using the biomformat R package, but keep getting errors related to number of columns.
Yes, that's exactly the table I was looking for. And it confirms that only ~10% of your reads are being kept, and also shows us which step is removing them.
And yet, quality remains low... How frustrating!
Having you tried trimming for shorter reads? Say 100 bp or 90 bp? Are you trimming at all at the start of the read?
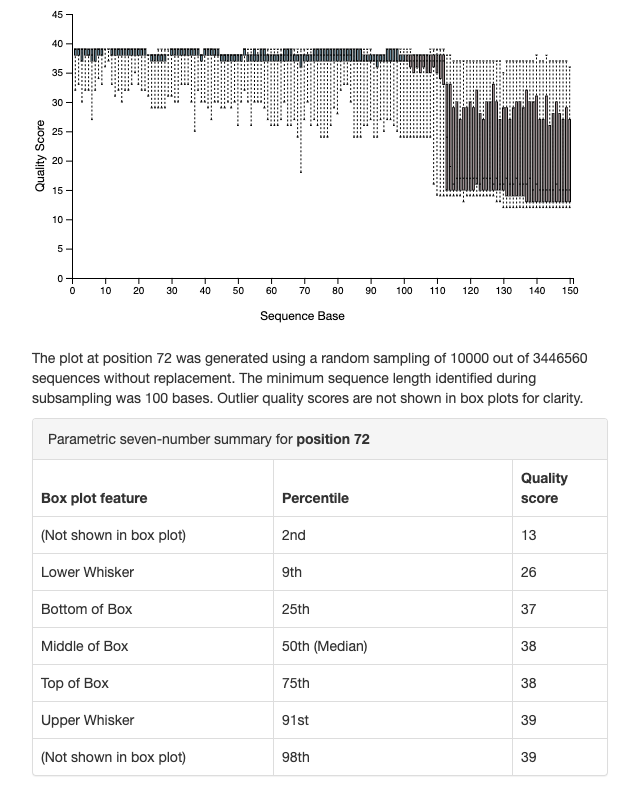
OK perfect! base 112 looks like a good choice based on those plots.
Dada2 can be picky about quality. Have you tried truncating at 110, just to make sure you don’t include any poor base pairs? Trimming at 100 could keep your quality near Q39, which is great, should also keep more reads.
Let me know if these other setting work well for you!
Thank you for the comment and I will definitely try that. But one thing that concerns me is that because my reads are too short, if my calculation is correct, my reads only cover around 50bp of the V3 region. Do you think such short coverage can provide useful information about the data?



{kind=link}