Dear All,
I´m running for the first time a dataset and would appreciated some help choosing the ideal parameters for DADA2.
Considering the quality control what trimming parameters should I choose for paired-end read analysis commands? It is clear from the graph that after 120 bases the quality begings to decrease...
Thanks in advance,
Francisco
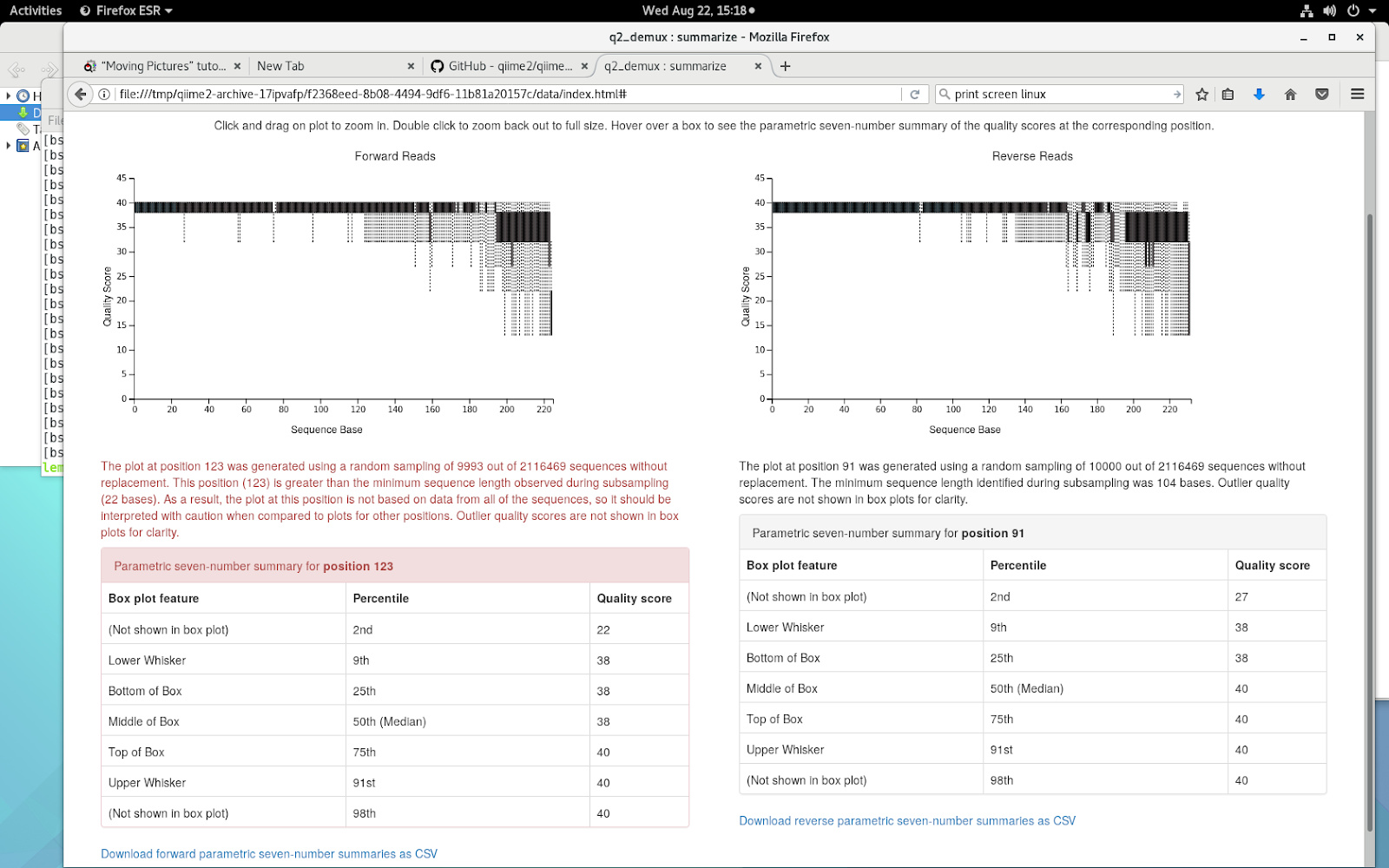
Hi @FranciscoC,
This is a common enough question around here so you could try searching the forum to see what others used based on their experience.
For paired-end reads we need to make sure that the sum of truncating parameters across the Forward and Reverse reads is not so much that it would leave insufficient overlap between the reads. DADA2 requires a min of 20bp overlap for proper merging, so you’ll want to calculate your overlap based on your specific primers and make sure you leave at least 20bp + natural variation of your expected target.
The quality plots you posted look fairly good and you can probably retain around ~200bp from each direction without losing too many reads. Another consideration for DADA2 is you want to make sure you’ve not done any prior quality control/trimming as the error-model building stage of DADA2 relies on the whole data set. I bring this up because your quality plots look a bit too straight and clean and typically we see more variability in the q-scores, ex. from Atacama soil tutorial, at least from Illumina reads anyways.
1 Like
Dear Mehrbod_Estaki, thank you very much for the guidance. I´m going to check if any quality control was done. If so, would Deblur be the appropriate option?
Again, many thanks,
Francisco
Hi @FranciscoC,
That will depend on what kind of pre-processing (if any) has been done. So let’s see what we can find out about those reads. To note, where DADA2 and Deblur differ in this regard is that DADA2 builds an error model based on the quality scores of your reads, while deblur will use a pre-packaged model which means it won’t need to use the actual quality scores.
1 Like
Dear Mehrbood,
Thank you very much for answering back. I have checked with the sequencing provider and they have removed the primers and barcodes, but no quality control was done. Considering this I have used the following parameters:
qiime dada2 denoise-paired
--i-demultiplexed-seqs demux.qza
--p-trim-left-f 0
--p-trim-left-r 0
--p-trunc-len-f 200
--p-trunc-len-r 200
--o-table table.qza
--o-representative-sequences rep-seqs.qza
--o-denoising-stats denoising-stats.qza
I have attach the stats results.
stats.tsv (2.8 KB)
Sincerely,
Francisco
Hi @FranciscoC,
That's good, this is what we want. Let's see how this plays out.
I can't seem to see your earlier quality screen file you attached in the first message, and the stats.tsv file you've attached is unreadable. Could you share the actual stats.qzv instead please?
Yes I can share the original table. Thank you.
Franciscostats (1).tsv (1009 Bytes)
Hi @FranciscoC,
Thanks for sharing the stats file. In the future, it would much more helpful if you were to share the actual stats.qzv output as mentioned since this has the added benefit of letting others view the provenance tab which is very helpful for troubleshooting. Never the less, its looks as though your issue is stemming at the merging step where about only 1/10 of your sequences being merged. As I mentioned in my first comment, for paired-end reads there is a minimum 20bp overlap that is required for proper merging, otherwise you end up in a situation like this where you lose all those that can’t be merged.
Could you please share the original summary visualizationa artifact with the quality plots (demux.qzv if you’re following the tutorials). The image link is broken in your original post.
Also, what is the primer set/target region you are using here? I’m guessing truncating at 200 in both directions is leaving insufficient overlap.
2 Likes
Hi @Mehrbod_Estaki,
Thank you for answering back. Attach you can find the original demux.qzv file. We are using the 515F-806R combination.demux.qzv (288.3 KB)
Thanks for sharing that @FranciscoC. So since you are using the short V4 region which has a long overlap section I don’t think this is the issue. The next step would be for us to make sure the amplification and sequencing was successful. If you were to upgrade your qiime2 version to 2018-6, the demux action will provide you with a nice summary of your reads’ length distribution. This will tell us if your reads are indeed long enough to properly overlap or not.
